Journal de Pharmacie Clinique
MENUNormes de préparation de produits stériles en pharmacie au Québec Volume 38, numéro 4, Décembre 2019

Illustrations
Tableaux
Contexte
Depuis de nombreuses années, le domaine pharmaceutique s’est grandement complexifié et plusieurs classes thérapeutiques de médicaments (p. ex. : antibiothérapie et chimiothérapie injectables) requièrent une préparation dans un environnement contrôlé pour assurer la qualité des produits et la sécurité constantes des patients. Dans le domaine pédiatrique entre autre, plusieurs médicaments font encore l’objet de préparations magistrales, notamment les produits stériles, en raison du manque de formulations commerciales adaptées à cette population. Cette situation a incité l’Ordre des pharmaciens du Québec (OPQ) à publier en 2014 deux normes actualisant celle de 1995 [1]. La norme 2014.01 concerne la préparation de produits stériles non dangereux [2] et la norme 2014.02, celle des produits stériles dangereux [3], autant pour le milieu communautaire que pour le milieu hospitalier. Ces deux normes miroirs formulent des conditions à respecter ainsi que des règles de bonne pratique favorisant des préparations exemptes de tous contaminants afin de maintenir la qualité des produits. La norme 2014.02 aborde également la protection du personnel contre les risques d’exposition chimique liés aux produits dangereux, de la réception du médicament jusqu’à son administration.
Les départements de pharmacie ont l’obligation de se conformer aux exigences de ces deux normes, notamment en élaborant des politiques et procédures sur les différents aspects de la préparation de produits stériles non dangereux et dangereux. Pour aider les départements de pharmacie des établissements de santé à se conformer à ces normes, l’Association des pharmaciens des établissements de santé du Québec (A.P.E.S.), une organisation formée en vertu de la loi sur les syndicats professionnels, a créé le Groupe de travail sur les préparations stériles (GTPS) en 2011. Le principal mandat du GTPS est de mettre à la disposition des départements de pharmacie des modèles de politiques et procédures et des outils qui tiennent compte des exigences des deux normes.
Objectif
Le présent document offre un aperçu des deux normes de pratique concernant la préparation de produits stériles en pharmacie au Québec. L’attention sera dirigée vers le processus de préparation aseptique, l’habillement, la méthode de détermination de la date limite d’utilisation des produits stériles (DLU) ainsi que la formation et l’évaluation du personnel.
Résultats
Les deux normes de l’OPQ s’inspirent du chapitre <797> de l’United States Pharmacopeia-National Formulary (USP-NF) en vigueur aux États-Unis depuis 2004 [4]. Elles s’appuient également sur l’avis d’un groupe de travail sur les préparations magistrales stériles et non stériles de l’Ordre des pharmaciens du Québec. Entre la norme précédente de 1995 et celles de 2014, un document intitulé « Conditions requises pour la préparation des produits stériles en pharmacie » a étépublié en 2010 et a servi de document de transition [5]. Les normes et le document sur les conditions requises ont occasionné des rénovations majeures des zones contrôlées des départements de pharmacie des établissements de santé au Québec. En 2013, le ministère de la Santé et des Services sociaux (MSSS) du Québec, qui veille au maintien, à l’amélioration et à la restauration de la santé des Québécois, a jugé bon de produire son propre document « Aires réservées aux préparations stériles (Unité de pharmacie) », afin de préciser plusieurs paramètres de construction [6].
En octobre 2017, l’OPQ a ajouté et publié un addenda aux deux normes de 2014. L’un des points principaux concerne l’ajout d’une section portant sur la DLU lors de toute modification apportée aux conditions d’entreposage.
Il faut rappeler que, au Québec, le système de distribution des médicaments est basé largement sur le concept de l’unidose (dose unique), avec une personnalisation de la production. Ce système de distribution nominative sur 24 heures de médicaments, tant stériles que non stériles, implique un service au nom du patient pour 24 heures à la fois, ce qui nécessite que la préparation soit faite le jour même ou la veille de l’administration. Si certaines préparations en lots sont effectuées à l’avance, elles ne sont pas moins servies individuellement. C’est d’ailleurs l’une des exigences normatives de l’OPQ, avec la constitution du dossier patient. La fabrication industrielle de produits stériles en vrac est soumise aux exigences de Santé Canada, qui est une instance fédérale. Seules les préparations stériles, individuelles ou en lots, destinées aux patients sont réglementées par l’OPQ, provincialement.
Le tableau 1 fait état des sections présentes dans les deux normes québécoises et dont plusieurs sont discutées dans cet article.
Notions générales tirées des normes 2014.01 et 2014.02 de l’OPQ pour les locaux réservés à la préparation des produits stériles
Salle blanche
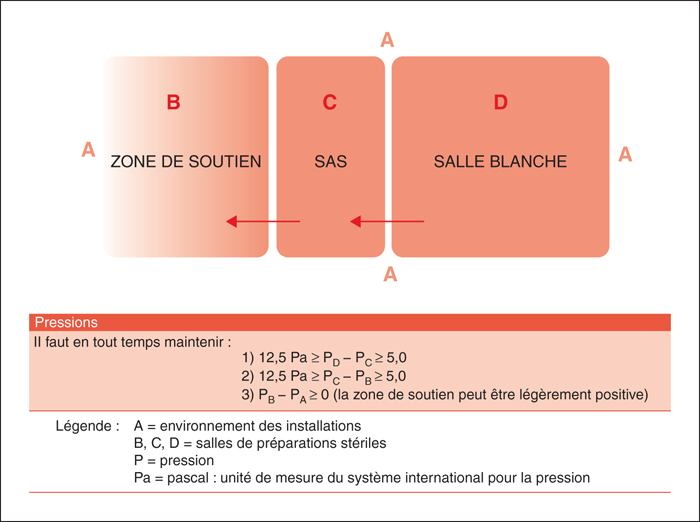
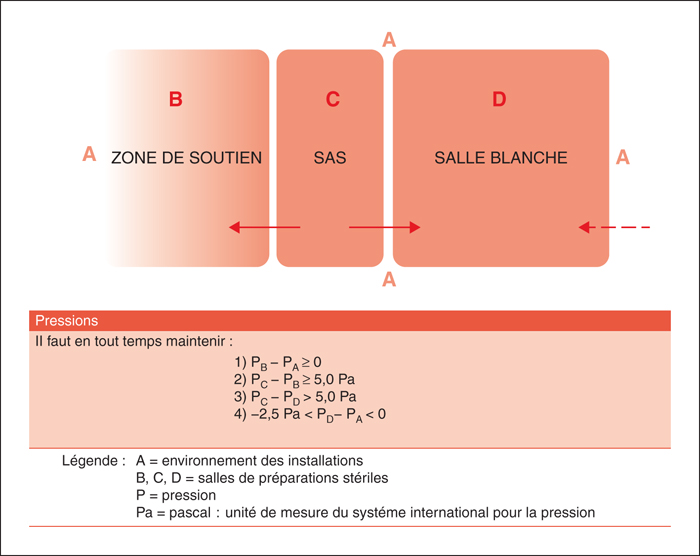
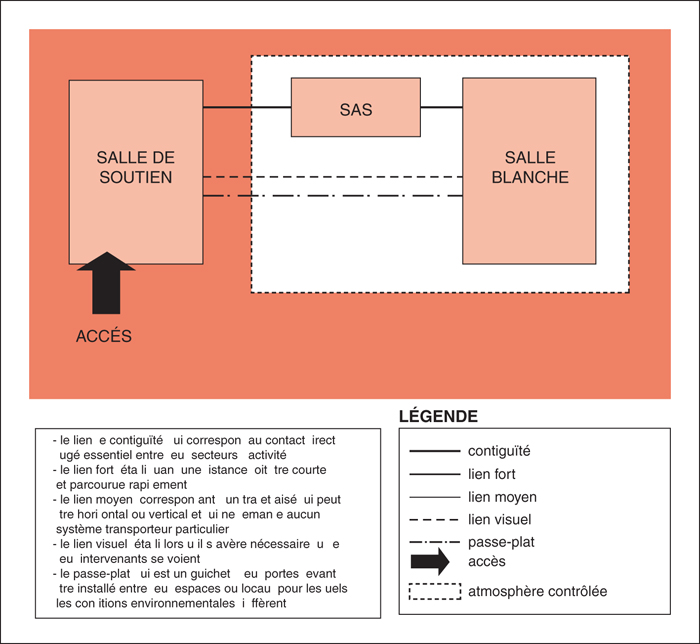
La salle blanche est une pièce étanche dans laquelle les propriétés de l’atmosphère (température, hygrométrie, teneur en particules et en micro-organismes, pression et mouvement de l’air, etc.) sont contrôlées. Les paramètres fonctionnels de la salle sont maintenus à un niveau précis. Les différents paramètres visés pour la salle blanche sont décrits dans le tableau 2 (produits stériles non dangereux) et le tableau 3 (produits stériles dangereux). Elle est conçue de façon à̀ réduire le plus possible l’introduction, la génération et la rétention de particules à l’intérieur [2, 3].
Sas
Le sas est l’enceinte ou le passage clos. Il est muni de deux portes dont le système de fermeture permet de n’ouvrir qu’une porte à la fois pour donner accès à la zone de soutien ou à la salle blanche, en maintenant ces milieux isolés l’un de l’autre. Les différents paramètres visés pour le sas sont décrits dans le tableau 4 (produits stériles non dangereux) et le tableau 3 (produits stériles dangereux).
Selon la norme 2014.01 (produits non dangereux), le sas est habituellement séparé en deux espaces délimités visuellement [2, 3] :
- –zone dite « microbiologiquement propre », attenante à la salle blanche de préparation de produits stériles ;
- –zone dite « microbiologiquement souillée » attenante à la zone de soutien de préparation de produits stériles.
Selon la norme 2014.02 (produits dangereux), le sas est aussi divisé en deux sections :
- –zone dite « microbiologiquement propre », avoisinant la zone souillée d’un côté et la salle blanche de l’autre. Même si cette zone est dite « propre », lorsque des produits dangereux sont préparés dans la salle blanche, elle est dite « contaminée » chimiquement ;
- –zone dite « microbiologiquement souillée », située à l’entrée du sas, dans la section avoisinant la zone de soutien. Même si cette zone est dite « souillée », lorsque des produits dangereux sont préparés dans la salle blanche, elle est dite « propre » chimiquement.
Zone de soutien
Attenante au sas, la zone de soutien est un espace réservé, ou une salle fermée selon les besoins, qui sert aux activités de soutien à la préparation de produits stériles [2, 3].
Les différentiels de pressions demandés sont indiqués sur la figure 1 (produits stériles non dangereux) et la figure 2 (produits stériles dangereux). La répartition générale des locaux est représentée sur la figure 3 (produits stériles non dangereux et dangereux).
Processus de préparation aseptique et habillement
Cette section expose les principes généraux qu’invoquait l’OPQ en émettant les deux normes québécoises de 2014, portant sur le processus de préparation aseptique et l’équipement de protection individuel (EPI) requis. Elle inclut les spécifications associées à la préparation des produits stériles dangereux.
Hygiène des mains, des avant-bras et habillement
Afin de maintenir le plus haut niveau d’asepsie possible, l’hygiène des mains, des avant-bras, ainsi que l’habillement, doivent se faire selon une séquence précise qui est détaillée dans les politiques et procédures de l’établissement.
L’EPI nécessaire à la préparation de produits stériles non dangereux comprend des couvre-chaussures, un bonnet qui couvre complètement les cheveux, un couvre-barbe (le cas échéant), un masque de procédure de type « chirurgical » pour protéger le produit, une blouse protectrice propre (faite de matériel à faible émission de particules et ajustée au cou et aux poignets), une paire de gants stériles non poudrés [2]. L’EPI se revêt et se retire dans la zone « microbiologiquement propre » du sas, attenante à la salle blanche.
L’EPI nécessaire à la préparation de produits stériles dangereux comprend certaines modalités supplémentaires et se revêt dans la zone propre chimiquement à l’entrée du sas [3, 7]. Le préparateur doit enfiler deux paires de gants et les changer régulièrement lors des activités de préparation se déroulant dans la salle blanche. Les poignets de la première paire doivent remonter sous les manches de la blouse et les poignets de la deuxième viennent par-dessus [3]. Il doit aussi porter une blouse protectrice imperméable, spécialement conçue pour la chimiothérapie. Ce vêtement est jetable et non réutilisable. En ce qui a trait à l’habillement lors du nettoyage de l’enceinte de sécurité biologique (ESB), les préparateurs doivent porter en outre « une visière de sécurité couvrant le visage ou des lunettes de sécurité combinées à un masque à cartouche [3] ». Une fois la préparation terminée, l’EPI doit être retiré dans la zone contaminée chimiquement du sas, selon la description rigoureuse des étapes définies dans les politiques et procédures [3, 7].
Introduction des produits et du matériel dans la salle blanche
Avant d’introduire les produits et le matériel dans la salle blanche, le personnel doit retirer les emballages cartonnés pour éviter la génération de particules. Seuls les emballages propres peuvent pénétrer dans cette zone contrôlée. De plus, les produits et le matériel doivent être préalablement désinfectés avec des lingettes ou compresses stériles, faibles en résidus et imbibées d’alcool isopropylique (AIP) à 70 % [2, 3]. Par la suite, l’ensemble du matériel désinfecté doit être déposé dans un récipient en plastique ou en acier inoxydable pour permettre son passage de la zone de soutien, ou du sas, vers la salle blanche.
Nettoyage et désinfection de l’enceinte de préparation stérile (EPS) ou de l’enceinte de sécurité biologique (ESB)
Lors du nettoyage et de la désinfection de l’enceinte, le personnel doit respecter certaines conditions. D’abord, avant d’entreprendre le processus, les préparateurs doivent se désinfecter les mains gantées avec de l’AIP à 70 % qu’ils laissent sécher ensuite. Puis ils doivent changer les compresses de nettoyage imbibées d’eau stérile à la suite de chaque section nettoyée. Cette étape est suivie d’une désinfection à l’aide de compresses stériles imbibées d’AIP à 70 %. L’EPS et l’ESB doivent être nettoyés au moins une fois par jour et désinfectés deux fois par jour, soit au début et à la fin de la journée de travail. Le personnel doit éviter en tout temps d’entrer la tête et le haut du corps dans l’enceinte de préparation [2, 3].
Décontamination et désactivation supplémentaires de l’ESB
Puisque l’AIP à 70 % ne possède pas les propriétés permettant d’inactiver les produits dangereux, le risque de contamination chimique subsiste. Une décontamination quotidienne de surface doit donc précéder l’étape de désinfection. Elle s’effectue avec un mélange d’eau et de détergent, par exemple un ammonium quaternaire, et se termine par un rinçage à l’eau stérile [3]. Le travail de désactivation avec de l’hypochlorite de sodium à 2,4 %, « suivi d’une neutralisation par le thiosulfate de sodium à 1 % est effectué une fois par mois et lors d’un déversement important ou si on soupçonne une contamination [3] », il est toujours suivi d’un lavage à l’eau stérile et d’une désinfection à l’alcool. Le tout selon la séquence suivante : désactivation (10 minutes), neutralisation (1 minute), rinçage et désinfection.
Vérification des préparations
Pour s’assurer que le processus de préparation aseptique soit réalisé adéquatement, un pharmacien ou son délégataire doit procéder à une vérification de manière à valider les ingrédients employés, les quantités mesurées et les techniques de manipulation exécutées par le personnel. La vérification peut se faire par observation directe, à travers une baie d’observation si elle permet la lecture précise du nom des ingrédients ainsi que des quantités mesurées ou par l’intermédiaire d’une caméra numérique avec ou sans prise de photo [2, 3].
Méthode de détermination de la date limite d’utilisation (DLU) des produits stériles
Date limite d’utilisation (DLU)
La norme 95.01 de l’OPQ était peu explicite au sujet de la détermination de la date de péremption des produits stériles comparativement aux nouvelles normes en vigueur. Pourtant, la détermination de la DLU s’avère une étape essentielle lors de la réalisation d’une préparation stérile. Elle fournit aux dispensateurs des informations cruciales, telles que la date et l’heure au-delà de laquelle il n’est plus possible de garantir la qualité de la préparation et, par conséquent, cette dernière n’est plus utilisable. D’autre part, la DLU varie en fonction des températures de conservation. « Lorsqu’aucune épreuve de stérilité spécifique n’est effectuée pour une préparation ou pour un lot, le pharmacien désigné au soutien des préparations de produits stériles doit établir une DLU selon les critères ci-dessous ».
Deux critères à considérer pour la DLU
La DLU ne doit pas dépasser la date la plus rapprochéedéterminée selon les deux critères suivants :
- –la date de péremption est basée sur la stabilité physico-chimique, selon les ouvrages de référence ;
- –la durée de conservation est liée au risque de contamination microbienne (la stabilité “microbiologique” liée au processus de préparation de produits stériles) [2, 3] ».
La DLU d’une préparation stérile s’établit arbitrairement en fonction du niveau de risque de contamination microbienne lors de la réalisation de la préparation et de la présence ou non d’agent de conservation dans la fiole.
DLU de la préparation stérile finale
La DLU de la préparation stérile finale dépend (tableau 5) :
- –du nombre d’heures s’étant écoulées entre la perforation initiale du septum de la fiole et son utilisation ;
- –de la complexité de la préparation (nombre d’unités stériles utilisées) ;
- –du mode de conservation (température ambiante contrôlée, réfrigérateur ou congélateur).
La méthode de détermination de la DLU des deux normes de l’OPQ s’inspire de l’USP-NF, raison pour laquelle on y trouve plusieurs similarités. Elle est déterminée à partir du moment où la préparation est achevée.
Niveau de risque de contamination microbienne
La prolifération bactérienne s’intensifie exponentiellement en présence d’un milieu nutritif propice en quelques heures seulement après la contamination. Il est nécessaire de tenir compte du risque de contamination microbienne lors de la détermination de la DLU, tel que l’illustre le tableau 6. La qualité de l’air de l’environnement où sont effectuées les préparations stériles influence le niveau de risque de contamination. À la pharmacie, les préparations doivent être réalisées à l’intérieur d’une EPS ou d’une ESB qui maintient au moins une qualité d’air de classe ISO 5. Cette EPS ou ESB se situe dans une salle blanche de norme ISO 7 adjacente à un sas de norme ISO 8 pour une EPS [2] et ISO 7 pour une ESB [3]. La norme ISO renseigne sur la qualité de l’air comme noté dans le tableau 7. Plus la valeur de la norme ISO est basse, plus l’air est dépourvu de particules.
Pour la classification selon le niveau de risque de contamination microbienne, il faut considérer le nombre d’unités stériles manipulées. Une unité stérile correspond à un contenant (p. ex. : une fiole, une ampoule, un sac de soluté, etc.). Voir plus de détails dans le tableau 6.
Les préparations à risque élevé de contamination contiennent au moins un ingrédient non stérile ou pouvant l’être. Elles doivent toujours passer par un processus de stérilisation. Divers processus existent, mais le plus utilisé et le plus accessible en milieu hospitalier québécois est la stérilisation par filtration membranaire. De plus, certains établissements utilisent la stérilisation par chaleur ou par gaz et très rarement aux rayons gamma.
Type de contenant du produit utilisé (unidose ou multidose)
Selon les normes québécoises, l’utilisation de produits stériles disponibles commercialement doit être privilégiée à la base. Un pharmacien ne devrait en aucune circonstance réaliser une préparation stérile à partir de composés non stériles uniquement dans le but de réduire les coûts [2, 3].
Le médicament provenant d’une ampoule doit être utilisé dès l’ouverture. On parle ici d’utilisation immédiate [2].
La DLU du contenu d’une fiole sans agent de conservation est de 24 heures au maximum après la première perforation du septum, qu’elle soit conservée à température ambiante contrôlée ou au réfrigérateur [2].
Pour une fiole utilisée dans les six heures après la première perforation du septum, le niveau de risque de contamination microbienne (risque faible, modéré, élevé) doit être évalué lors de la détermination de la DLU pour la fabrication d’un lot [2].
Pour une fiole unidose (sans agent de conservation) utilisée plus de six heures après la première perforation du septum, la DLU de la préparation finale est de 24 heures à la température ambiante contrôlée et de 48 heures au réfrigérateur, indépendamment du niveau de risque de contamination microbienne de la préparation, du nombre d’unités stériles utilisées ou de sa complexité. Il est à noter que des lots ne peuvent en aucun cas être préparés à partir de cette fiole [2].
La DLU de la fiole multidose est de 28 jours après la première perforation du septum, à moins d’avis contraire du fabricant [2-4].
Préparations dans les situations urgentes à court terme
Dans les cas où le cadre normatif ne peut être entièrement respecté, il est possible de procéder à la réalisation de préparations stériles de qualité pour un usage à court terme. Néanmoins, certaines conditions s’appliquent.
Préparation pour un usage immédiat dans les situations urgentes (réalisée hors zone contrôlée)
Pour la préparation réservée aux situations urgentes et immédiates, les conditions à respecter sont les suivantes :
- –la préparation ne dépasse pas trois “unités stériles” ;
- –la préparation ne contient aucun médicament dangereux (p. ex. : chimiothérapie) ;
- –il y a deux perforations ou moins dans le site d’injection d’un produit sans agent de conservation pour chaque unité stérile utilisée ;
- –la technique aseptique ne requiert pas plus d’une heure de préparation en continu ;
- –la technique aseptique est rigoureusement respectée ;
- –la préparation est effectuée dans une situation d’urgence où l’administration immédiate au patient est requise [2].
Si toutes les conditions sont respectées, la DLU d’une préparation pour usage extemporané est d’une heure, qu’elle soit conservée à température ambiante ou au réfrigérateur.
Préparation avec des DLU de 12 heures au maximum dans les situations urgentes
Les préparations avec des DLU de 12 heures au maximum concernent les préparations réalisées dans une enceinte au moins de classe ISO 5, mais située dans un environnement de qualité inférieure à ISO 7. L’autorisation de réaliser des préparations dans ce contexte n’est accordée que pour des situations provisoires. Les conditions à respecter sont les suivantes :
- –les préparations sont de faibles risques seulement ;
- –les préparations ne contiennent aucun produit dangereux ;
- –une seule préparation est effectuée à la fois ;
- –les préparations sont effectuées dans un endroit réservé à la préparation de produits stériles et qui minimise le risque de contamination ;
- –il n’y a pas d’évier dans cet endroit, pas de fenêtres non scellées, pas de porte donnant sur l’extérieur ou dans une zone où la circulation est élevée, l’endroit est non adjacent à des sites de construction, des entrepôts ou encore des lieux où l’on prépare de la nourriture [2].
Situation particulière
Pour la préparation en lots de seringues d’insuline pour une administration sous-cutanée, il n’est pas obligatoire, selon la norme OPQ 2014.01, de l’exécuter en milieu stérile sous une EPS si certaines conditions et restrictions sont étroitement respectées. Il est à noter que cette particularité québécoise n’existe nulle part ailleurs, à notre connaissance, et n’est inscrite que pour des raisons pratiques à l’exercice de la pharmacie en milieu communautaire. La DLU pour ce type de préparation est établie à neuf jours au réfrigérateur. Se référer au point 7.13 « Situation particulière : mise en seringue d’un produit pour une administration sous-cutanée » de la norme 2014.01 [2].
Changement de condition d’entreposage
Dans l’addenda d’octobre 2017, l’OPQ ajoute à ses normes de 2014 le point 7.1.5 « DLU applicable lors d’un changement de condition d’entreposage ». Cette section précise que les « DLU aux différentes conditions d’entreposage ne sont pas cumulatives et le temps d’entreposage total d’une préparation de produits stériles ne doit jamais dépasser la DLU initiale (microbiologique ou physico-chimique, la plus courte des deux) [2, 3] ».
Pour illustrer le propos, prenons l’exemple d’une préparation conservée au réfrigérateur, possédant une DLU initiale de 14 jours, qui est retirée du réfrigérateur le 13e jour et laissée par la suite à la température ambiante. Si nous supposons une DLU à la température ambiante de 48 heures, la nouvelle DLU ne pourrait pas dépasser 24 heures.
Pour conclure, le début d’administration d’une préparation stérile doit se faire avant la DLU mentionnée.
Formation et évaluation du personnel
Lors de l’exécution d’une préparation stérile, le risque de contamination dépend entre autres des techniques aseptiques du manipulateur. Un programme de formation et d’évaluation des compétences du personnel s’avère donc essentiel et obligatoire [2, 3, 8].
En plus de la formation académique préalable à l’embauche, le personnel assigné à la préparation de produits stériles doit suivre un programme initial de formation donné par le département de pharmacie. Ce programme doit inclure la lecture et la compréhension des politiques et procédures, une formation théorique, une formation pratique individualisée, une évaluation théorique, une évaluation pratique par observation des techniques aseptiques, un échantillonnage des bouts de doigts gantés, un test de remplissage aseptique et un examen théorique. Une fois le programme de formation suivi avec succès, le manipulateur, assistant technique ou pharmacien, obtient le droit de commencer à préparer des produits [2, 3, 8].
Ensuite, un programme d’évaluation du maintien des compétences du personnel doit prendre le relais. Il comprend un test pratique et théorique. Le personnel doit être évalué au moins annuellement en ce qui a trait aux préparations comportant un risque faible et modéré et aux préparations de produits dangereux. Quant aux préparations comportant un risque élevé, une évaluation semestrielle est nécessaire, soit deux fois par an. Le personnel manipulateur qui échoue perd son droit d’exercer ses fonctions rattachées aux préparations stériles et doit reprendre le processus de formation [2].
Bien que le personnel du département de pharmacie ou de la pharmacie communautaire ne soit pas assigné aux tâches d’hygiène et de salubrité, l’OPQ précise qu’il incombe au pharmacien désigné de s’assurer que l’entretien soit effectué adéquatement.
Politiques et procédures
Les normes de l’OPQ imposent un cadre réglementaire pertinent pour la réalisation de préparations stériles sans toutefois expliquer concrètement la façon de procéder. Pour préciser les impératifs des normes et leur mode d’application, la rédaction de politiques et procédures est indispensable à la description de certaines obligations et sont exigées par les normes de l’OPQ. Ces politiques et procédures doivent être approuvées par le pharmacien responsable des produits stériles dans l’établissement et il lui appartient de les gérer et de les faire appliquer. Elles décrivent notamment toutes les nuances et les démarches supplémentaires devant être prises pour la gestion des médicaments dangereux, ce que la norme 2014.02 détaille peu.
Le GTPS de l’A.P.E.S. a ainsi élaboré des modèles de politiques et procédures et des outils pour soutenir les activités de ses membres qui autrement auraient tous eu à les rédiger chacun de leur côté. Cinquante et un documents ont été écrits relativement aux préparations de produits stériles non dangereux et cinquante-quatre pour les préparations de produits stériles dangereux, sans compter la boîte à outils, les vidéos et la procédure d’évaluation du pharmacien désigné au soutien. Les membres de l’A.P.E.S. peuvent y accéder sur le site Web de l’Association dans une section non publique. Ces outils sont modifiables à la discrétion des établissements de santé, sans que ce soit une obligation pour eux de le faire. Il s’agit plutôt de recommandations qui simplifient grandement le travail du pharmacien responsable, qui n’a alors qu’à les adapter à ses besoins.
Conclusion
Les deux normes de l’OPQ portant sur les préparations stériles fournissent au personnel des départements de pharmacie des cadres minimaux à respecter pour optimiser les conditions d’asepsie. L’objectif ultime consiste à offrir aux patients une préparation sans contaminant, permettant une administration sécuritaire et de qualité élevée. Par conséquent, la rigueur est essentielle lors des contrôles de qualité. Pour terminer, il est intéressant de noter que l’Association nationale des organismes de réglementation de la pharmacie (ANORP) du Canada a adapté les normes de l’OPQ afin de les proposer aux ordres professionnels des autres provinces canadiennes.
Remerciements
Nous tenons à remercier Sandrine Gobeil, candidate au programme de Pharm.D. au moment de la rédaction du document, ainsi qu’Alexandre Lagacé et Jennifer Farrah, pharmaciens, pour leur contribution à la relecture de l’article.
Liens d’intérêts
les auteurs déclarent ne pas avoir de lien d’intérêts en rapport avec cet article.
 Cette œuvre est mise à disposition selon les termes de la
Licence Creative Commons Attribution - Pas d'Utilisation Commerciale - Pas de Modification 4.0 International
Cette œuvre est mise à disposition selon les termes de la
Licence Creative Commons Attribution - Pas d'Utilisation Commerciale - Pas de Modification 4.0 International