Hépato-Gastro & Oncologie Digestive
MENUPolykystoses rénales et polykystoses hépatiques Volume 26, numéro 8, Octobre 2019

Introduction
Les polykystoses des reins et du foie forment un groupe hétérogène de maladies génétiques à transmission autosomique dominante, caractérisées par le développement progressif de kystes multiples dans les deux reins et le foie. Schématiquement, la polykystose hépatique (PKH) est le plus souvent associée à une polykystose rénale à transmission dominante (PKRAD) – on parle de polykystose hépato-rénale. Bien plus rarement la PKH est isolée, sans kystes rénaux. L’imagerie échographique est la pierre angulaire du diagnostic : dans le contexte de PKRAD familiale, la détection avant 40 ans de > 3 kystes répartis dans l’un ou les deux reins suffit à établir le diagnostic chez un individu à risque pour la maladie [1]. Le diagnostic de polykystose hépatique repose conventionnellement sur le constat de > 20 kystes dans le foie, un seuil qu’un groupe d’experts a récemment proposé d’abaisser à la présence de > 10 kystes hépatiques [2]. Polykystose rénale à transmission dominante et polykystose hépatique isolée sont des maladies génétiques à transmission dominante
Épidémiologie et génétique
Polykystose rénale autosomique dominante
La polykystose rénale à transmission dominante (PKRAD) est une néphropathie tubulo-interstitielle chronique, responsable d’une insuffisance rénale progressive. La maladie expose en outre à deux manifestations extra-rénales fréquentes, une polykystose hépatique (90 % des patients) et les anévrismes cérébraux (environ 10 %). La PKRAD se singularise par son évolution lente, et des conséquences rénales qui surviennent à l’âge adulte, le plus souvent après 30 ans.
La prévalence de la PKRAD oscille entre 1/1 000 et 1/2 500. La maladie est décrite dans toutes les populations. Elle vient en fréquence au premier rang des maladies rénales héréditaires. En France, elle est responsable d’environ 9 % des causes d’insuffisance rénale terminale : en 2017, 7 % des 47 000 patients dialysés et 13 % des 38 000 greffés de rein avaient une PKRAD. La sévérité de la maladie rénale est tantôt similaire, tantôt un peu plus marquée chez les hommes que chez les femmes [3].
La PKRAD résulte habituellement d’une mutation dans PKD1 (en France, 78 % des familles, chromosome 16) ou PKD2 (15 % des familles, chromosome 4) [4]. Récemment, deux autres gènes ont été identifiés dans un tout petit nombre de familles, GANAB et DNAJB11 [5, 6]. Le gène muté reste inconnu dans 5 % des familles PKRAD. Les mutations des deux gènes PKD1 et PKD2 ont une pénétrance complète dans la quasi-totalité des cas. La survenue d’une mutation de novo concerne environ 5 % des patients PKRAD, et une mosaïque, germinale ou somatique peut être prouvée chez < 1 % des patients. Environ 1 500 mutations différentes sont connues dans PKD1, et 250 dans PKD2 [4].
Polykystose hépatique isolée
C’est une maladie ultra-rare (incidence, 1/100 000). L’hétérogénéité génétique est importante, puisque six gènes distincts ont été incriminés (PKRCSH, SEC63, LRP5, GANAB, SEC61B et ALG8) dans 25-50 % des PKH isolées, mais la signature moléculaire reste inconnue dans plus de la moitié des familles [7]. La pénétrance de l’atteinte hépatique est très faible [8] et le phénotype va donc d’une maladie asymptomatique à une polykystose hépatique massive.
Cette revue laisse délibérément de côté les autres affections génétiques où coexistent une polykystose rénale et une atteinte hépatique non-kystique, notamment :
- –La forme récessive de polykystose rénale liée aux mutations de PKHD1 : les anomalies biliaires consistent chez les homozygotes en une dilatation segmentaire des voies biliaires intrahépatiques associée à une fibrose hépatique ; des kystes du foie peuvent être observés chez les hétérozygotes qui sont en revanche dépourvus de kystes des reins [3].
- –Une forme exceptionnelle d’hyperinsulinisme par mutation du promoteur de PMM2 où des kystes hépatiques sont possibles [9].
- –La maladie par mutation de HNF1β où les reins sont de taille diminuée et les kystes rénaux peu nombreux, et associés à des anomalies fluctuantes des tests hépatiques sans kyste hépatique [10].
Physiopathologie des kystes des reins et du foie
Les gènes PKD1 et PKD2 codent pour les protéines transmembranaires polycystine-1 (PC1) et polycystine-2 (PC2) exprimées dans le cil primaire des cellules tubulaires rénales et des cholangiocytes. Le cil primaire est une antenne cellulaire unique présente au pôle apical des cellules épithéliales et susceptible de capter des signaux mécaniques ou biochimiques, et d’activer des voies de signalisation intracellulaire. PC1 agit comme mécano-récepteur de surface cellulaire sensible au flux urinaire. PC2 est également localisée dans le réticulum endoplasmique ; elle appartient à une famille de canaux calciques non sélectifs. Les deux protéines PC1 et PC2 interagissent par leurs extrémités C-terminales. Cette interaction est déterminante pour la maturation de PC1, son adressage au cil primaire et sa stabilisation. Couplées, les deux protéines régulent la concentration de calcium intracellulaire. À l’exception de LRP5, les produits des gènes mutés dans la PKH isolée sont impliqués dans le contrôle de qualité de la maturation des glycoprotéines exercé par le réticulum endoplasmique, en particulier au bénéfice de PC1. LRP5 interagit avec les protéines transmembranaires Frizzled à la membrane cellulaire. L’implication étroite du cil primaire dans la physiopathologie de la PKRAD et de la PKH a abouti à proposer que ces maladies soient classées dans le vaste cadre des ciliopathies [11]. Polycystine-1 et polycystine-2 sont des protéines ciliaires
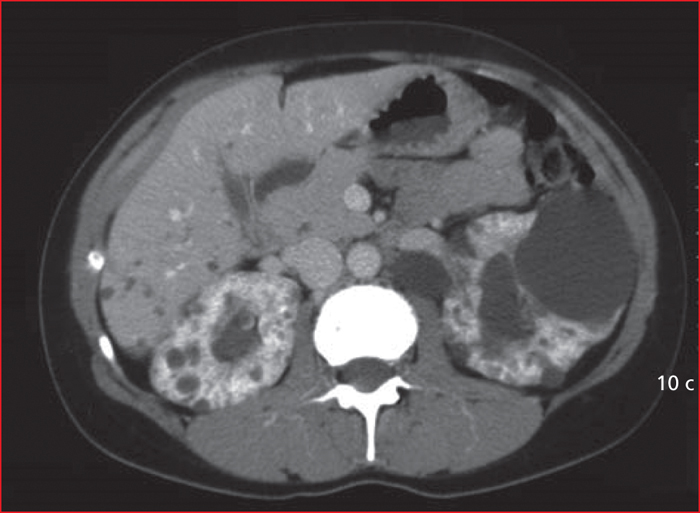
En résumé, les mutations des gènes impliqués dans la PKRAD ou la PKH isolée aboutissent à une production défaillante, une maturation insuffisante ou un défaut d’adressage de PC1 (ou de PC2). La kystogenèse rénale ou hépatique survient lorsque l’activité fonctionnelle de PC1/PC2 s’abaisse en deçà d’un seuil. Quoique les fonctions précises des deux protéines PC1 et PC2 restent incertaines, le déficit en activité PC1 (ou PC2) aboutit à une baisse de concentration intracellulaire du calcium, d’où s’ensuivent un excès d’AMPc, et l’activation de voies intracellulaires impliquées dans la prolifération cellulaire (dont mTor, MAPK, Wnt et β-caténine) et la sécrétion du fluide kystique. Les kystes des reins dérivent de tous les segments du néphron, y compris le tube collecteur. Les kystes du foie sont attribués à une résorption incomplète de la plaque ductale, et à la persistance de dilatations segmentaires des cellules biliaires primitives. Chez les patients ayant hérité d’une variation génétique pathogène de PKD1 ou PKD2 ou d’une forme génétique de PKH, le développement des kystes est focal comme illustré sur lafigure 1, alors que toutes les cellules portent un allèle muté : à l’âge adulte, la transformation kystique ne concerne qu’une fraction (1-10 %) de l’ensemble des néphrons (ou des cholangiocytes). Parmi les mécanismes responsables de cette kystogenèse focale, la survenue d’une deuxième mutation (second hit) a été postulée et prouvée dans la PKRAD et dans la PKH isolée : la perte d’hétérozygotie peut intéresser l’allèle sauvage du gène, ou un gène impliqué dans le réseau des gènes kystiques (cystogènes) : on parle alors de transhétérozygotie [12]. Les protéines transmembranaires polycystine-1 et 2 contrôlent le calcium et l’AMPc intracellulaires, et en aval la kystogenèse
Complications rénales de la polykystose rénale à transmission dominante
Les maladies liées à PKD1 et à PKD2 sont similaires, avec des kystes rénaux innombrables et une augmentation progressive du volume des reins, mais l’impact rénal est plus précoce et plus marqué dans les familles PKD1. Chez l’adulte jeune, les reins sont plus volumineux que dans les familles liées à PKD2, et ultérieurement l’âge médian de l’insuffisance rénale terminale est de 58 ans (PKD1) vs. 79 ans (PKD2)[3]. La polykystose rénale due aux mutations de GANAB se distingue par un petit nombre de kystes rénaux, et la préservation de fonction rénale [5]. Enfin, sans les familles mutées pour DNAJB1, les reins sont de taille normale ou diminuée, avec une évolution tardive possible vers l’insuffisance rénale terminale [6].
Hypertension artérielle
C’est une manifestation fréquente et précoce de la PKRAD : la moitié des patients sont hypertendus dès 20-30 ans, donc bien avant le début de l’insuffisance rénale. L’HTA s’accompagne d’une stimulation du système rénine-angiotensine. L’objectif est de maintenir la pression artérielle < 140/90 mmHg. Si un régime limité en sel (6 g/j) et les modifications du style de vie ne suffisent pas, le traitement de première ligne est un inhibiteur de l’enzyme de conversion. L’intérêt et la faisabilité d’un contrôle plus strict de la pression artérielle (< 110/75 mmHg) chez le patient PKRAD avec une fonction rénale bien préservée avant 50 ans (débit de filtration glomérulaire > 60 mL/min/L, 1,73 m2) [13] sont débattus [14].
Douleurs lombaires et abdominales
Une pesanteur lombaire chronique uni- ou bilatérale est une manifestation omniprésente des gros reins polykystiques. Une douleur aiguë unilatérale survient chez 60 à 80 % des patients, avec trois possibilités diagnostiques :
- –Le plus souvent une hémorragie intra-kystique spontanée, qui est un accident très fréquent dans la PKRAD. La douleur est fixe, l’hémorragie est attribuée à la rupture d’un vaisseau de la paroi kystique ; elle peut donner lieu à une hématurie, si l’hémorragie concerne un kyste profond proche de la voie excrétrice, ou à un hématome sous-capsulaire ou péri-rénal si le kyste hémorragique est périphérique, cortical. Le patient est habituellement apyrétique mais une fièvre (< 38,5 ̊C) est possible si le saignement est abondant. L’échographie peut visualiser un kyste au contenu hétérogène, la tomodensitométrie (TDM) (figure 2) visualise un kyste hyperdense (densité spontanée > 50 UH). Repos (au lit) et antalgiques sont indiqués, les symptômes régressent en 2 à 7 jours.
- –Une migration lithiasique (incidence cumulée, environ 20 %) : la douleur est une colique néphrétique sans fièvre (en l’absence de rétention purulente), les cavités rénales sont dilatées en échographie, la TDM est le meilleur examen pour localiser le calcul qui est constitué d’acide urique ou d’oxalate de calcium (hypocitraturie). Les techniques de lithotriptie extra-corporelle, ou de traitement endo-urologique des lithiases s’appliquent à la PKRAD. La prévention repose sur les apports hydriques abondants, et une alcalinisation par bicarbonate de sodium ou citrate tri-potassique en cas de lithiase uratique.
- –Une infection rénale, qui doit être suspectée de principe si la douleur est fébrile ou a été précédée de signes de cystite. L’infection du rein droit doit être soigneusement distinguée d’une complication hépatique (Encadré 1). Le rein polykystique infecté ressort de deux causes possibles, pyélonéphrite ascendante (l’ECBU documente la bactériurie) ou infection de kyste (l’ECBU identifie une pyurie aseptique). Cette dernière doit être suspectée si la CRP est > 70 et que la TDM ne détecte pas d’hémorragie intra-kystique. Le PET-scan peut être utile pour localiser le kyste infecté, le diagnostic est établi par l’analyse du fluide kystique (pus et entérobactéries). Le traitement combine drainage per-cutané et antibiothérapie pendant quatre semaines, initialement en association (fluoroquinolone ou céphalosporine de troisième génération et aminoside).
L’insuffisance rénale
C’est la complication grave la plus fréquente de la PKRAD. La fonction rénale reste longtemps préservée dans la maladie, avant qu’une diminution progressive de la filtration glomérulaire débute entre 30 et 60 ans. La perte de fonction rénale est en moyenne de – 5 mL/min/an. Comme mentionné plus haut, les patients mutés pour PKD1 progressent plus rapidement vers l’insuffisance rénale terminale : celle-ci survient selon les séries à un âge médian de 50-55 ans, et chez les patients PKD2 entre 69 et 80 ans. De plus, dans les familles liées à PKD1, l’impact des mutations tronquantes est plus sévère que celui des mutations non-tronquantes (âge médian de l’insuffisance rénale terminale : 55 ans vs. 67) [3]. À cette variabilité allélique se superpose une variabilité intrafamiliale fréquente dans la PKRAD illustrée par l’âge de survenue de l’insuffisance rénale terminale chez les individus d’une même famille, qui peut notablement diverger (jusqu’à 20 années), suggérant l’implication de facteurs d’environnement ou de gènes modificateurs, dont certains sont des allèles de cystogènes mutés et cohérités [3]. Le volume rénal et un score clinico-génétique prédisent le risque d’insuffisance rénale terminale
Deux approches ont été développées pour prédire le pronostic rénal à long terme : la mesure du volume rénal, et un score clinico-génétique.
Le volume rénal rapporté à la taille prédit le pronostic rénal
Les reins augmentent progressivement de volume au fil des années dans la PKRAD. Le volume rénal total (VRT) peut être mesuré après un scanner ou une imagerie par résonance magnétique (IRM) à l’aide de logiciels de traitement d’images. Une estimation fiable du VRT peut aussi être obtenue en routine à l’aide de la formule ellipsoïde (π/6 × hauteur × épaisseur × diamètre transverse) en utilisant la valeur maximale de chacune des trois dimensions de chacun des deux reins. L’étude CRISP, et les travaux dérivés qui tiennent compte de la taille des patients, ont établi la capacité du rapport VRT/taille à prédire la vitesse de déclin rénal en fonction de l’âge au moment de la mesure [14, 15] : un score (Mayo Imaging Classification) permet d’attribuer un risque individuel de progression lente (classes 1A ou 1B) ou rapide (classes 1C, 1D ou 1E) de l’insuffisance rénale. Les agences sanitaires américaine et européenne ont reconnu le bien-fondé de cette approche, et décidé de considérer le VRT comme un marqueur intermédiaire acceptable dans l’évaluation des traitements spécifiques opposables à la PKRAD.
Un score mixte clinique et génétique prédit le pronostic rénal
Un algorithme pronostique (PRO-PKD) a été développé en France par l’équipe de Le Meur et al., qui attribue un score à des marqueurs cliniques simples (sexe, HTA avant 35 ans, complication urologique avant 35 ans [hématurie, ou douleur rénale ou infection de kyste]) et à la variété de mutation familiale (PKD1 tronquante, PKD1 non-tronquante, ou PKD2). Ce score prédit avec acuité un risque faible, modéré ou élevé de progression vers l’insuffisance rénale terminale à 60 ans [3].
Diagnostic
Comment et quand faire le diagnostic de polykystose rénale à transmission dominante ?
Le diagnostic de PKRAD repose sur deux critères : l’histoire familiale et l’imagerie. L’histoire familiale (établir un arbre généalogique) est dite positive si l’anamnèse établit la notion d’une néphropathie kystique avec transmission dominante chez un parent (ou un enfant) du propositus. L’imagerie de première intention est l’échographie. Chez un sujet à risque, l’aspect caractéristique est celui de deux reins augmentés de taille, dont les contours sont déformés par des kystes innombrables. À un stade plus précoce, les reins sont le siège d’un petit nombre de kystes corticaux et médullaires, et ne sont pas augmentés de taille.
L’âge doit être pris en considération, et dans les cas litigieux, les critères échographiques validés pour établir le diagnostic de PKRAD en présence d’une histoire familiale positive sont les suivants [1] : entre 15 et 39 ans, > 3 kystes répartis dans un ou les deux reins ; entre 40 et 59 ans, > 2 kystes dans chacun des reins ; à 60 ans ou plus, > 4 kystes dans chacun des reins. Après l’âge de 40 ans, le constat de 0-1 kyste exclut la maladie. Entre 30 et 39 ans, l’absence de kyste exclut l’existence de la maladie à 99 %.
La TDM injectée et l’IRM sont plus sensibles que l’échographie pour détecter les kystes des reins (dès 2-3 mm), et les dénombrer. L’une ou l’autre technique est indiquée si l’échographie est douteuse et qu’un diagnostic formel est indispensable rapidement : candidat à un don de rein pour une greffe rénale familiale, ou sujet jeune qui souhaite disposer de cette information pour son avenir. Les critères mentionnés plus haut pour le diagnostic échographique de PKRAD diffèrent pour la TDM ou l’IRM.
En l’absence d’histoire familiale de PKRAD, le diagnostic peut être retenu si les deux reins sont polykystiques et augmentés de taille, et associés à une polykystose hépatique.
À quel âge réaliser une échographie abdominale chez un mineur asymptomatique « à risque » ?
En l’absence de traitement spécifique avant 18 ans, une conférence de consensus récente a laissé ouverte les deux options : différer le dépistage au-delà de la majorité ou réaliser une échographie de dépistage à intervalle régulier (tous les trois ans) si l’exploration initiale est négative [16].
Traitement ciblé de la polykystose rénale à transmission dominante
Les essais thérapeutiques infructueux
Un nombre significatif d’essais cliniques ciblant diverses voies de signalisation ont été entrepris ou sont en cours dans la PKRAD. Les inhibiteurs de mTor (éverolimus, sirolimus et rapamycine), les analogues de la somatostatine (qui diminuent l’AMPc), un inhibiteur de l’HMG-CoA réductase et un inhibiteur de tyrosine kinase (bosutinib) ont échoué à montrer un effet bénéfique sur le volume rénal ou la pente de déclin de la fonction rénale. Sont en cours d’évaluation la metformine (un stimulateur de l’AMPK, AMP-activated proteine kinase) et le venglustat (un inhibiteur de la glucosylceramide synthase) et, plus simplement, un apport hydrique abondant (inhibant la sécrétion de vasopressine qui stimule l’AMPc).
Le tolvaptan, unique traitement approuvé
Le tolvaptan est un antagoniste spécifique des récepteurs V2 de la vasopressine (l’hormone anti-diurétique) dont l’expression est restreinte aux cellules principales du tube collecteur dans le néphron distal. Ce produit a été testé initialement chez des individus à risque élevé de progression rénale (VRT > 750 mL avant 50 ans avec clairance de créatinine > 60 mL/min/1,73 m2) dans l’essai randomisé TEMPO 3:4, où son efficacité a été établie : à une posologie de 60-120 mg/j, le médicament réduit (objectif principal) significativement la vitesse de croissance annuelle du volume rénal total (+2,8 % vs. +5,5 % dans le groupe contrôle) et (objectifs secondaires) ralentit le déclin de la fonction rénale. Enfin, il réduit les douleurs liées à la maladie [17]. Cet essai a suffi pour obtenir une autorisation de l’EMA, alors que la FDA a exigé une étude complémentaire dont l’objectif principal concernait l’impact sur le débit de filtration glomérulaire. Cette étude (REPRISE) a établi dans une frange plus vaste de patients (DFG entre 90 et 45 mL/min) que la pente annuelle du DFG était significativement infléchie par le tolvaptan (–2,34 vs. –3,6 mL/min/an) [18]. L’effet aquarétique prononcé du médicament est un obstacle à son utilisation régulière. Une toxicité hépatique chez 2-3 % des patients traités nécessite une surveillance mensuelle des tests hépatiques pendant 18 mois. Si ces précautions et une contraception efficace sont acceptées par les patients, le médicament peut être prescrit et renouvelé par un néphrologue aux conditions suivantes : PKRAD à risque élevé de progression et donc un VRT > 600 mL/m ou une dimension du rein > 16,7 cm, un DFG > 25 mL/min/1,73 m2, et des signes d’évolution rapide cliniques (ceux du PRO-PKD score, voir ci-dessus) ou une baisse du DFG > 3,5 mL/min/an. Les recommandations européennes ou nord-américaines publiées depuis les essais pivots ont insisté sur la nécessité de ne pas entreprendre le traitement après 55 ans, faute d’évaluation dans cette classe d’âge [19, 20]. Le tolvaptan ralentit le déclin rénal dans la polykystose rénale à transmission dominante
Traitements de l’insuffisance rénale terminale
Dialyse (hémodialyse ou dialyse péritonéale) et transplantation rénale (avec donneur vivant ou non) peuvent être proposées pour le traitement de suppléance rénale. La dialyse péritonéale est possible, sauf en cas de très gros reins. La greffe de rein est considérée comme le traitement optimal [4], et peut être envisagée jusqu’à 75 voire 80 ans en l’absence de comorbidités. En préparation à la greffe, une néphrectomie (ou éventuellement une réduction du volume rénal par embolisation radiologique) peut être indiquée pour libérer l’espace nécessaire pour implantation du greffon, ou se débarrasser d’un (ou deux) rein(s) ayant donné lieu à des complications infectieuses ou hémorragiques récidivantes.
Polykystose hépatique
Facteurs de risque
Dans la PKH isolée, la pénétrance est faible (< 5 %) quel que soit le gène muté [4]. Au cours de la PKRAD, des kystes hépatiques sont détectables chez > 90 % des patients âgés de plus de 35 ans [14] et une atteinte hépatique symptomatique concerne environ 20 % des patients [21]. Plus de 80 % des patients symptomatiques sont des femmes. La croissance moyenne du volume hépatique est d’environ 1-2 % par an, mais nettement différente chez les femmes avant et après 48 ans (+ 4,8 % vs. + 0,6 %) [22]. L’impact des estrogènes de synthèse sur la croissance du foie polykystique est débattu. Chez les femmes ayant une forme modérée ou sévère de polykystose hépatique, l’usage d’une contraception incluant un œstrogène et le traitement hormonal substitutif sont vivement déconseillés. L’usage d’estrogènes à visée contraceptive ou substitutive est contre-indiqué dans la polykystose hépatique modérée ou sévère
Volumétrie hépatique
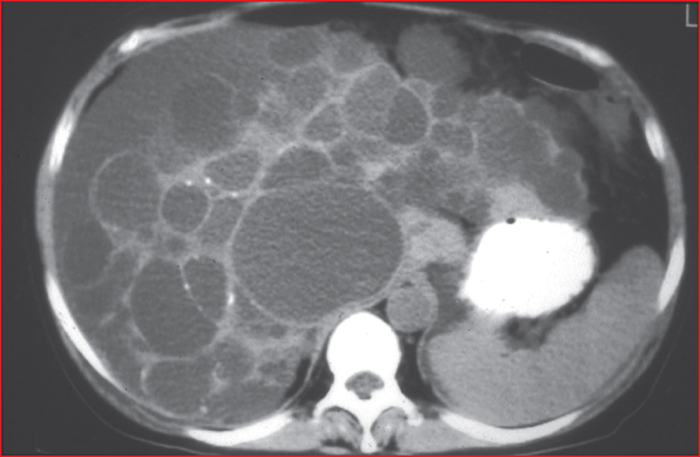
L’atteinte hépatique se résume le plus souvent à une hépatomégalie asymptomatique, avec un foie bosselé et des tests hépatiques normaux. Les symptômes éventuels de la PKH incluent pesanteur chronique et douleurs, satiété précoce, perte d’appétit et nausées. Leur sévérité peut être évaluée à l’aide d’un questionnaire [23]. À l’instar de l’imagerie rénale et du VRT, la mesure du volume hépatique (rapportée à la taille) permet une appréciation complémentaire de l’ampleur volumétrique de la PKH qui est arbitrairement classée en minime (volume < 1 600 mL/m, le plus souvent asymptomatique), modérée (1 600 à 3 200 mL/m), ou massive (> 3 200 mL/m) [24], avec un impact sur la qualité de vie [23].
Complications de la polykystose hépatique
Elles sont de deux ordres, les complications focales et les complications liées à l’hépatomégalie diffuse.
Complications focales
La douleur élective : c’est par exemple le cas de kystes (> 4 cm diamètre) dont la localisation est mal tolérée (notamment sous le rebord costal ou à l’épigastre) [25].
L’hémorragie intrakystique est très fréquente, avec des symptômes brefs (kyste de petite taille) ou intenses et durables si le kyste concerné est de grande taille (> 8 cm). Au scanner, la densité d’un kyste hémorragique est supérieure à 50 UH. Le traitement symptomatique des douleurs est toujours préférable à un geste percutané ou chirurgical.
L’infection de kyste hépatique (incidence : 0,1 % patient/an) donne un tableau de fièvre avec douleur localisée du foie, et sur la TDM (ou l’écho) un kyste au contenu hétérogène avec paroi épaissie. Le germe responsable est le plus souvent une entérobactérie. Le PET-scan est indiqué pour localiser le(s) kyste(s) infecté(s) si le scanner n’est pas contributif. Un drainage est indiqué si l’antibiothérapie isolée échoue à traiter l’infection [26](Encadré 1).
Complications des hépatomégalies massives
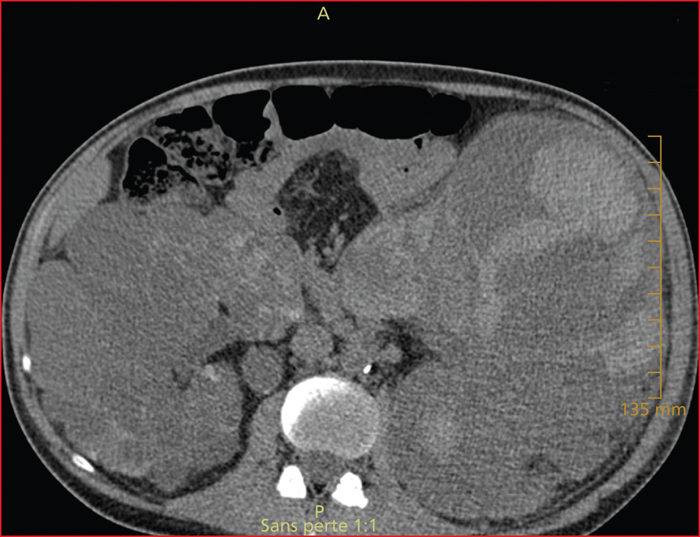
L’hépatomégalie modérée ou massive retentit sur les organes de voisinage et peut provoquer un syndrome de petit estomac avec satiété rapide, un pyrosis, des troubles du transit, une dénutrition avec amaigrissement et sarcopénie, une dyspnée (due au refoulement thoracique ou à la compression des cavités du cœur droit) et des œdèmes (compression cave inférieure). La compression des veines sus-hépatiques par les kystes peut être responsable d’une obstruction sus-hépatique compliquée d’ascite réfractaire (figure 3)[25]. Une augmentation de la gamma-GT et de la phosphatase alcaline est habituelle. Il n’y a jamais d’insuffisance hépato-cellulaire [25]. L’hépatomégalie massive, qui altère la qualité de vie et le schéma corporel, a souvent une contrepartie psychologique.
La tomodensitométrie injectée est la meilleure imagerie pour évaluer la PKH symptomatique : sont à considérer la taille et la diffusion des kystes, la compression des veines hépatiques, l’existence d’une circulation collatérale, la présence d’ascite, et la déperdition musculaire (psoas). Diverses classifications de la PKH ont été proposées pour standardiser la prise en charge thérapeutique, dont la plus usitée est celle de Chigot [21].
Options thérapeutiques et leurs indications dans la polykystose hépatique symptomatique modérée ou massive
Analogues longue durée de la somatostatine (lanréotide ou octréotide)
Ils réduisent l’AMPc et le calcium intracellulaire, et réduisent la sécrétion du fluide kystique et la prolifération des cellules bordant les kystes. Ils ont un effet sur les symptômes et le volume hépatique, avec une réponse hétérogène, mais plusieurs essais randomisés ont établi une réduction modérée ou une stabilisation du volume hépatique sur une période brève ou semi-prolongée (6-36 mois), et une amélioration de la qualité de vie [27-30]. La tolérance globale est bonne, en dehors de troubles digestifs à l’initiation du traitement. L’effet d’un traitement prolongé peut être durable, mais un échappement est possible (20 % des patients au-delà de deux ans). Les analogues de la somatostatine n’ont pas d’impact favorable ou délétère sur la fonction rénale [30]. Un analogue de la somatostatine peut réduire ou stabiliser le volume hépatique
Ponction-aspiration et sclérose
Cette approche peut être proposée pour le traitement de douleurs focales chroniques en rapport avec un kyste de 4 à 7 cm de diamètre. L’aspiration du liquide kystique suivie de l’injection d’un agent sclérosant sous contrôle radiologique est efficace dans deux tiers des cas, avec une récurrence tardive chez 20 % des patients. La procédure peut être répétée [21, 22, 25].
Fenestration des kystes
Elle peut être proposée aux patients se présentant avec plusieurs kystes dominants dans les segments antérieurs du foie, et donc accessibles à une laparoscopie. L’amélioration symptomatique est effective chez 90 % des patients. Une ascite post-opératoire est fréquente, et souvent réfractaire si coexiste une insuffisance rénale chronique (DFG < 30 mL/min). Une récidive symptomatique est observée dans 25 % des cas [22].
Résection hépatique
Elle est réservée à un petit nombre de patients sélectionnés dans les rares centres familiers de cette approche, car la morbi-mortalité est élevée. Les kystes modifient considérablement l’organisation vasculaire et biliaire du foie rencontrée avec des difficultés chirurgicales per-opératoires considérables, et les adhésions post-opératoires compliquent une éventuelle greffe hépatique ultérieure. La régénération hépatique post-opératoire ne met pas à l’abri d’une récidive éventuelle de PKH symptomatique [21, 31].
Transplantation hépatique
La greffe d’un foie est l’option préférentielle en cas de retentissement marquée d’une PKH massive, du fait de complications locales (infections de kyste récidivant ou hypertension portale avec ascite) ou systémiques (dénutrition, perte d’autonomie). La mortalité précoce oscille entre 5 et 10 %, la survie du patient et du greffon à cinq ans approche 90 % [2, 31]. Une greffe combinée foie et rein doit être considérée si le DFG est < 40 mL/min [25].
Conclusion
La PKH est en principe une maladie bénigne, mais dans les formes massives une prise en charge doit être concertée. Le traitement de la PKH doit être restreint aux patients symptomatiques. Le choix entre les options de traitement pharmacologique, radiologique ou chirurgical est guidé par quelques repères simples incluant l’intensité des symptômes, la diffusion intra-hépatique des kystes (localisation, taille, nombre) et l’état général. Dans le futur, les efforts devront tendre dans deux directions : identifier des biomarqueurs (imagerie ou biologie) susceptibles de prédire précocement les patients à risque de développer une PKH invalidante ; et identifier des voies thérapeutiques innovantes.Take home messages
Liens d’intérêts
L’auteur a participé comme investigateur associé aux deux essais mentionnés dans les références 17 et 18. Interventions ponctuelles : Otsuka.
 Cette œuvre est mise à disposition selon les termes de la
Licence Creative Commons Attribution - Pas d'Utilisation Commerciale - Pas de Modification 4.0 International
Cette œuvre est mise à disposition selon les termes de la
Licence Creative Commons Attribution - Pas d'Utilisation Commerciale - Pas de Modification 4.0 International