Epileptic Disorders
MENUThe phenotype and treatment of SCN2A-related developmental and epileptic encephalopathy Volume 22, numéro 5, October 2020

- Mots-clés : SCN2A, developmental and epileptic encephalopathy, sodium channel blockers
- DOI : 10.1684/epd.2020.1199
- Page(s) : 563-70
- Année de parution : 2020
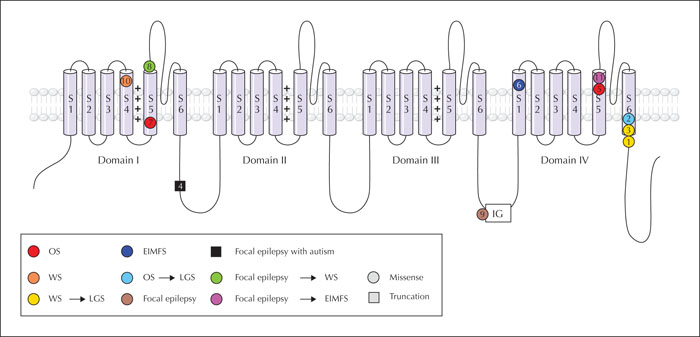
Aims. We aimed to delineate the phenotypic spectrum of SCN2A-related developmental and epileptic encephalopathy (DEE) and determine the effectiveness of various treatment modalities, including sodium channel blockers and the ketogenic diet.
Methods. Eleven patients with SCN2A-related DEE were included in the study. The characteristics of SCN2A mutations, electroclinical features, clinical course, and response to treatment modalities were analysed.
Results. The 11 patients were aged between 0.4 and 9.7 years. The onset of seizures ranged from neonate (six patients) to infant (four patients), to childhood (one patient). Epilepsy presented as Ohtahara syndrome, West syndrome, epilepsy of infancy with migrating focal seizures (EIMFS), and focal epilepsy in neonatal- to infantile-onset patients. The only childhood-onset patient in our study presented with focal epilepsy with autism. Neonatal-to infantile-onset patients had drug-resistant epilepsy (9/10), however, sodium channel blockers were effective in all treated patients (9/9). The ketogenic diet (6/8) and high-dose steroid treatment (4/5) were also effective. The seizures in the childhood-onset patient worsened during treatment with sodium channel blockers. All mutations in neonatal- to infantile-onset patients were missense mutations, whereas the mutation in the childhood-onset patient was a truncation mutation.
Conclusions. These results support earlier observations regarding the epilepsy syndromes and response to antiepileptic drugs in patients with SCN2A-related DEE.