Epileptic Disorders
MENUMeasurable outcomes for pediatric epileptic encephalopathy: a single-center experience with corticosteroid therapy Volume 23, numéro 1, February 2021


Refractory epilepsy affects 2 per 1,000 children in the United States [1]. Despite an increasing number of anti-seizure medications, a third of children with epilepsy continue to have inadequate control of their seizures [2]. Corticosteroids are commonly used for refractory epilepsy [3, 4]. In specific epilepsy syndromes, steroid use is supported by randomized control trials (for West syndrome) [5-7] or meta-analyses of case series (for continuous spike waves in sleep) [8, 9]. However, outside of these specific syndromes, there are few studies to guide therapy [10-12].
Current evidence for corticosteroid treatment for epileptic encephalopathy consists primarily of small observational studies [10, 13]. In a Cochrane review, there was a single randomized control trial for corticosteroid use (ACTH) in childhood epilepsy other than West syndrome that included only three children in the final analysis [13]. Corticosteroids were found to be helpful for generalized seizure control in some, however, the ideal timing and patient that might benefit from corticosteroid treatment remains elusive [4].
The population currently treated with corticosteroids is heterogeneous and includes children with epileptic encephalopathy (EE). EE are syndromes in which epileptic activity adversely affects brain function over and above what is expected of the underlying etiology [14]. However, the etiologies, pathophysiology, and key clinical features are diverse even within a single named epileptic encephalopathy syndrome. For example, Lennox-Gastaut syndrome is a clinical phenotype that can result from several different etiologies, such as hypoxic-ischemic encephalopathy or cortical malformations [15].
It is also unclear if corticosteroids should be used to target clinical seizures, the EEG, or both. Steroids are also used to target the encephalopathy itself, though evidence to support this remains primarily based on expert opinion and clinical [9, 16, 17]. This gap in knowledge contributes to the uncertainty regarding which clinical setting steroids may be useful for, the aspect of epilepsy targeted by treatment, and the timing, dosing, and type of steroid to be used.
Addressing these gaps is particularly important because steroid treatment appears to be effective in a subset of patients. Better defining the subset in whom efficacy is seen is hindered by the heterogeneity and undefined treatment targets previously mentioned. In a review of 192 pediatric patients with refractory epilepsy, 48% responded to corticosteroid treatment (defined as >50% reduction in seizures) [12]. The effect size is much larger than expected from a third or four ASM [18] and comparable to response rates of non-pharmacological therapies such as a vagal nerve stimulator [19] or the ketogenic diet [20, 21]. Sustained improvement in seizures with corticosteroid treatment has been repeatedly reported [10, 22]. Given our limited arsenal of effective treatments for refractory epilepsy, corticosteroids should not be overlooked. Here ,we review our experience with corticosteroids in children with refractory epilepsy in order to identify domains for outcome measurement, characterize the heterogeneity in our treated population, and highlight knowledge gaps to be addressed in future studies.
Methods
This was a single-center retrospective cohort study to describe the effect of corticosteroids on children with refractory epilepsy. The institutional review board at Weill Cornell Medical Center (WCM) approved this study (IRB #1812019863).
Study population
We reviewed charts for all pediatric patients seen in the Weill Cornell Medicine refractory epilepsy clinic from January 2006 to December 2018. Individuals diagnosed with an epileptic encephalopathy that was not infantile spasms (categorized or not) and managed primarily at WCM who received either IV methylprednisolone or oral dexamethasone were included. Children were excluded for the following reasons:
- –over the age of 18;
- –received steroid therapy for a different indication (i.e., asthma);
- –concurrent use of another immunomodulatory agent (such as IVIG or anakinra);
- –and no outpatient epilepsy follow-up visit information was available.
Additionally, individuals were excluded if steroid course was altered due to:
- –extension of dexamethasone course by more than one week;
- –and/or a change in dosing (dexamethasone or methylprednisolone).
Data abstraction
One reviewer (JG, a board-certified pediatric epileptologist) abstracted the electronic medical record. Data abstraction included determination if corticosteroids (either IV or oral) were used to treat epilepsy, epilepsy history (including age at onset of epilepsy, epilepsy etiology, seizure types), seizure frequency, seizure duration, epilepsy syndrome type, and prior workup including imaging and genetic studies. Demographic data included age, sex, race, ethnicity, and insurance status. Epilepsy treatment history included current and prior medication use, use of the ketogenic diet or vagal nerve stimulator (VNS), and history of epilepsy surgery. Side effects from corticosteroid treatment were elicited at clinical visits and extracted from the medical record.
Corticosteroid treatment
We compared two differentstandardized corticosteroid regimens: IV methylprednisolone or oral dexamethasone; the two most commonly used regimens in our institution and elsewhere [12, 16, 23]. Corticosteroid treatment is routinely used at our institution for regular clinical management of refractory epilepsy. Patients were counseled regarding risks and side effects and provided verbal consent prior to treatment. Methylprednisolone was administered at a dose of 30mg/kg/day IV (max 1 g) daily for three days in the inpatient setting, concurrent with video EEG monitoring. Oral dexamethasone was administered in the outpatient setting with the following 10-day course: Day 1-2: 4 mg four times daily; Day 3-4: 4 mg three times daily; Day 5-6: 4 mg twice daily; Day 7-8: 4 mg daily; Day 9-10: 2 mg daily [24]. Selection of steroid course was at the discretion of the neurology provider, and was often guided by the psychosocial situation, ability to comply with the treatment protocol, and tolerability. All patients were counseled regarding side effects prior to therapy initiation and all were treated with ranitidine or a proton pump inhibitor.
Long-term video electroencephalography (EEG) assessments
We reviewed the raw traces of long-term video EEG monitoring. For each subject, there was a pre-treatment EEG and at least one post-treatment EEG. The EEG that was prior and closest to the time of steroid treatment was selected as baseline EEG. The initial post-treatment EEG was the EEG closest and after the final dose of steroid in a given course. In some subjects, an additional follow-up EEG (EEG #2) was obtained if the patient was monitored again without inter-current steroid treatment. One reviewer (JG) reviewed all EEG recordings. The reviewer was not blinded to the patient but was blinded to which EEG was pre or post-treatment as well as to the clinical EEG interpretation report.
Functional domain assessments
Parental assessment of improvement after steroid treatment was also recorded from the medical record. The following parental assessment domains were abstracted: overall improvement, improvement in seizure frequency and duration, improvement in motor skills, improvement in behavior, improvement in cognition, and improvement in sleep. Seizure frequency was tracked per parental preference and could include use of seizure diary or seizure app.
EEG analyses
For each individual, the baseline EEG was compared with both the post-steroid treatment EEG (EEG #1) and follow-up EEG if available (EEG #2). Timing of steroid effect was captured by use of two post-treatment EEGs whenever possible. Patients were only included in the analysis of the second follow-up EEG (EEG #2) if there were no additional intervening steroid treatment of any kind. It is important to note that individuals for whom EEG#2 was obtained may be biased toward those with a less dramatic response to corticosteroid treatment noted in EEG#1, as those with a clear response often received another dose of steroids before a second follow-up EEG was obtained. The following EEG assessments were made:
- –number and duration of seizures (electroclinical or electrographic) in the first 24 hours of recording;
- –rescue medication needed during the EEG (yes/no);
- –EEG organization;
- –epileptiform activity;
- –presence and description of slowing (table 1);
- –and presence and description of sleep spindles (none/rudimentary/yes).
Because an aim of this study is to inform future outcome measurements in corticosteroid trials, we also calculated the sample size needed based upon the effect size identified here.
The scales used to analyze background waking organization were novel. The five-point ordinal scale was adapted from the glossary of terms put forth by the International Federation of Clinical Neurophysiology [25] and the primary electrographic features highlighted by the International League Against Epilepsy in the development of the standardized computer-based organized reporting of EEG (SCORE) [26]. These features included assessment of frequency, amplitude, anterior-to-posterior gradient, and presence of a posterior dominant rhythm, as well as epileptiform activity (table 1). To estimate the inter-rater reliability of the ordinal scales, two pediatric epileptologists [KG and BOM] reviewed five of the EEG studies – each blinded to the details of the patient and to other reviewers’ scores.
Seizure assessments
Seizure assessments were aimed at capturing the key “domains of seizure burden” identified by parents of children with severe refractory epilepsy and defined by Berg et al[27].These domains included: seizure frequency, seizure severity and seizure unpredictability. Seizure frequency was captured both by manual electrographic seizure counts on pre and post-treatment EEGs. Parental assessments of seizure frequency improvement were also obtained but were not used for seizure counts, as obtaining accurate assessments of tonic seizure frequency based on reporting is challenging [28].Seizure counts were further examined by seizure subtype based upon the ILAE 2017 seizure classification; generalized motor, generalized non-motor, focal motor, focal non-motor, and unknown onset [29]. When possible, seizures were further classified into subtypes including tonic, myoclonic, epileptic spasms, atypical absence. Those individuals with a specific seizure subtype were compared prior to and post-treatment with corticosteroids. Seizure severity was captured by seizure duration and need for rescue medication. Seizure duration was measured in 10-second increments (smallest being a <10-second seizure) due to concern in accurately defining onset and offset. The need for rescue medication was recorded during the video-EEG admission from the medical record. Due to the retrospective nature of this study, seizure predictability was not able to be captured, however, parents were asked if seizure burden overall improved.
Initial treatment comparison analyses
Exploratory sub-analyses were performed to identify outcomes associated with type of corticosteroid used (methylprednisolone vs. dexamethasone). If patients had received both types of steroids at some point during their treatment course, only the initial treatment was included in analysis. Comparison between side effects, clinical seizure burden, EEG features, and parental assessments of efficacy was performed between the dexamethasone and methylprednisolone treated groups.
Statistical and online tools
Study data were collected and managed using REDCap (Research Electronic Data Capture) [30] hosted at Weill Cornell Medical College. We used STATA 15 (StataCorp) for Fisher's exact test and Wilcoxon rank sum tests. We used R (R Foundation for Statistical Computing) for intra-class correlation coefficients (ICC) as a measure of inter-reader reliability. ICC was tested with the kappa statistic and qualified as follows: poor <0; slight 0–0.20; fair 0.21–0.40; moderate 0.41–0.60; suboptimal 0.61–0.80; and almost perfect >0.80 [31, 32]. Power calculations were performed using http://www.statistics.com/content/javastat.html interactive statistical calculator based upon the effect sizes identified in this study to guide future clinical trials [33].
Results
Of the 388 patients screened, 39 patients met initial inclusion criteria. Of these, four had neither a follow-up EEG nor visit data available and thus were excluded from the final analysis. Thirty-five (22 male) subjects were included in the study and 38 had follow-up EEGs. Among those with follow-up EEGs (EEG #1), 16 received methylprednisolone and 19 received dexamethasone. A second follow-up EEG (EEG #2) was obtained in 13 patients (eight treated with methylprednisolone; five with dexamethasone). The first post-treatment EEGs occurred at a median of two [interquartile range: 0-14] days and the second (EEG#2) at 42 [30-60] days from the last day of the initial steroid course. Clinical visits were often independent of EEG visits and occurred at a median of 17 [14-29] days post-treatment.
Characterizing the heterogeneity of the population
Most individuals had structural acquired etiology (43%), followed closely by genetic (31%) (supplementary table 1). Epileptic encephalopathy diagnoses were varied; 16 were clinically diagnosed with LGS and four with late-infantile epileptic encephalopathy, as defined by Nordli et al. [34]. The remaining 15 did not carry a more specific diagnostic subtype of epileptic encephalopathy. Of these 15, 12 had tonic seizures as their predominate seizure type, two had Dravet syndrome and one had Doose syndrome. Patients were on a median number of three (IQR: 2-3) anti-seizure medications (ASMs) at the time of seizure treatment. The most frequently used ASMs were clobazam (n=18; 51%), followed by levetiracetam (n=17; 49%) and topiramate (n=16; 34%). Additionally, 13 (37%) were on the ketogenic diet at the time of treatment and 11 (31%) had a VNS in place. Median age at epilepsy onset was one (0-3) year. Median time from epilepsy diagnosis to first steroid treatment was five (2-9) years (table 2).
Identifying domains for outcome measurements and effect size
Seizure frequency recorded on EEG decreased in 15 individuals (45%) from baseline to EEG#1. There was no significant decrease in number of seizures captured on EEG between the pre-treatment EEG and EEG#1 (18 [9-26] vs. 13 [5-21]; p=0.32) or between baseline EEG and EEG#2 (23 [7-39] vs. 12 [-3-26] in 24 hours; p=0.30). One patient with LGS became seizure-free after a single course of IV methylprednisolone. None of the patients required seizure rescue medication during video-EEG recording.
The number of patients with at least one seizure captured on video-EEG was 28/35 (80%) at baseline and 27/35 patients (77%) on post-treatment EEG#1, and 7/13 (54%) on post-treatment EEG#2. The most frequent clinical seizure types seen included generalized motor (including myoclonic, tonic, clonic, tonic-clonic; n=27; 77%), of which tonic (n=26; 74%) and myoclonic (n=17; 49%) were the most frequent. There was a significant improvement in tonic seizure frequency (24-hour seizure counts pre-treatment from 8 [4-13] to 3 [1-5] at EEG#1; p=0.02). In total, 16/35 (46%) of individuals had a >50% reduction in seizure frequency from the pre-treatment EEG to EEG#1. Of these, six were treated with methylprednisolone and 10 with dexamethasone (38% vs. 53%; p=0.35). Seizure duration did not change using our crude measurement; the percentage of patients with seizures lasting <one minute improved from 91% (pre-treatment EEG) to 100% (post-treatment [#1 or #2]; p = 0.42). Given our effect size of nine percentage points, we calculated that a sample >140 individuals will be needed to identify a significant change in seizure duration in future studies.
At the first post-treatment EEG, there were no significant differences compared with the baseline EEG waking background organization, slowing or epileptiform activity (table 3). There was, however, improvement in the presence of sleep spindles in between their baseline and first post-treatment EEG; 13 individuals (37%) had normal sleep spindles noted pre-treatment and 22 individuals (63%) normal spindles on EEG#1 (p=0.04). Interestingly, when the baseline EEG was compared with the second follow-up EEG (EEG#2), in which a smaller number was examined, there was no significant difference in sleep architecture (37% normal to 46% normal; p=0.34). At the second follow-up EEG, there was a significant improvement in the ordinal organization scale (p=0.02) (table 3, figure 1), in that hypsarrhythmia was no longer seen (initially two had hypsarrhythmia) and more patients had limited or primarily organized background (23% to 46%). There was no significant correlation between years from diagnosis to treatment and improvement in epileptiform scale (spearman rho: 0.06, p=0.75 for EEG#1 and spearman rho: 0.08, p=0.80 for EEG#2) or organization (spearman rho: 0.09, p=0.63 for EEG #1, spearman rho: 0.45, p=0.12 for EEG #2).
Between the baseline EEG and the initial post-treatment EEG, there were five patients who had medication changes: two discontinued a medication (clonazepam, topiramate), one was weaned off a medication (levetiracetam), and two had additional medication (felbamate, clobazam). The ketogenic diet was not stopped for any individuals treated with corticosteroids.
Comparing the IV methylprednisolone treated group and the oral dexamethasone treated group, there were no significant differences in demographic factors at baseline (table 2), nor in EEG findings at either time point (table 2), nor parental report of seizure improvement (table 4). However, cognitive improvement was reported more frequently among patients who received dexamethasone over methylprednisolone (11/19 vs. 4/16; p=0.03).
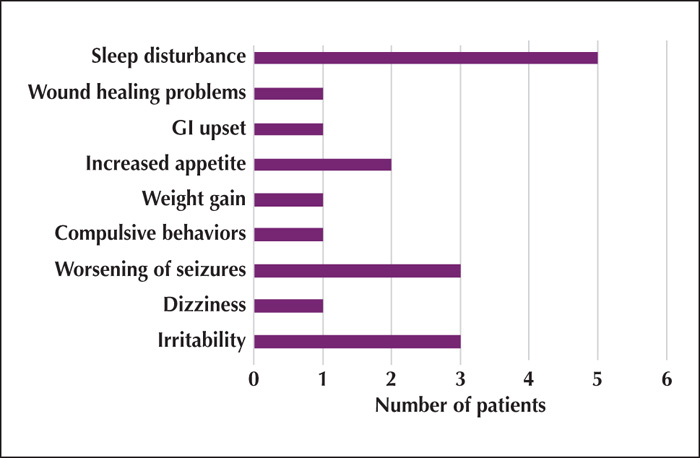
No patients stopped steroids early due to side effects. Side effects were reported in 14 (40%) of the 35 total subjects; the most common being sleep disturbance reported in 12 subjects (figure 2). There was no significant difference in side effects reported between the two regimens (p=0.62).
Identification of measurement gaps for future clinical trials
Good or excellent concordance among the EEG readers was present for all scales used to analyze the EEG, based on three reviewers (JG, KG, and BOM) for five EEGs (table 1). Epileptiform activity had a moderate intra-class correlation coefficient of 0.44. Intra-class correlation was suboptimal for ordinal scale measures of background (0.71) and almost perfect for slowing (0.88), and spindle activity (0.82) (p < 0.05 for all four measures).
Parental assessments were gathered from the first post-treatment visit after steroid therapy at a median 17 (IQR 14-29) days after steroid treatment (i.e., often in the outpatient setting prior to the next EEG). Most parents (62%) reported overall improvement from the steroid course (table 4). The improvement domain most frequently reported was an improvement in seizure frequency (n=18; 51%), followed by cognition (n=14; 40%) (table 4). For all 15 patients who had a reduction in EEG seizures from pre-treatment to post treatment, the parents also reported a decline. However, there were three patients in whom the seizure frequency was not reduced on EEG, but the parents reported improvement.
Discussion
Summary of findings
Single-course corticosteroid therapy was associated with a subsequent decrease in seizure frequency in nearly half (n=16; 46%) of this cohort. Tonic seizures were the most frequently identified seizure type and were significantly reduced from pretreatment to the first follow-up EEG (from eight [4-13] EEG seizures in the first 24-hour pre-treatment period to three [1-5] EEG seizures post-treatment). There was no clear benefit in seizure control using IV methylprednisolone vs oral dexamethasone, but some suggestion (from parental report) that oral dexamethasone resulted in better cognitive improvement. Sleep architecture was the only background improvement noted at the first follow-up EEG (median of two days), but this improvement was transient. Improvement in EEG organization was noted in the second post-treatment EEG (occurring a median of 42 days post-treatment), suggesting corticosteroid effects may not be immediate, although this finding is strongly limited by the number of second follow-up studies available. Taken together, out data suggest that future studies should be designed to capture reduction in seizure frequency, background organization, and sleep architecture changes. Assessments of cognitive change may be important outcome measurements to integrate into larger clinical studies.
Population heterogeneity
Etiology of epilepsy, known epilepsy syndrome and timing of treatment were diverse in this patient cohort. For example, timing of treatment ranged from 0 to 17 years from diagnosis. Thus, at our center, as in others, epileptologists are recommending corticosteroid treatments for individuals with both structural and non-structural etiologies, defined epilepsy phenotypes and poorly defined epilepsy syndromes, recently diagnosed epilepsy and long-standing epilepsy [16, 22]. Despite this heterogeneity, efficacy was still seen with single-course corticosteroid treatment. For example, 46% of individuals had a >50% reduction in seizures at EEG#1. This finding supports the use of corticosteroids for multiple types of epileptic encephalopathies.
We did not find an association between time from diagnosis to corticosteroid treatment and efficacy of treatment, however, all of these patients were already refractory and treated years after diagnosis (median of five years). Therefore, the full effects of corticosteroid treatment early in the disease course may not have been captured. Future studies will need to standardize timing of treatment to better capture the effect size of corticosteroid treatment.
Future domains to capture
Seizure burden, including frequency, severity and predictability, is an important outcome measure in refractory epilepsy, highlighted by both caretakers and physicians [27, 35].Our findings support seizure frequency as an important outcome measure in studies of corticosteroid therapy. Here, manual counting of seizures based on the video-EEG record illustrated a reduction in tonic seizures by the first time point, and overall seizure frequency in a selected cohort at a later time point (second post-treatment EEG). Severity (delineated by seizure duration and need for rescue medication) did not show clear improvement in this cohort. This may be secondary to the crude measures of seizure severity; for example, most patients (91%) had seizures lasting <one minute at baseline, and using this measure, a sample of >133 individuals would be needed to show an effect if the reduction seen here is real. Prior studies have identified a lack of seizure predictability as an important measure of seizure burden which was not captured here but should be included in future studies [27].
Clinical implications
Tonic seizures, in particular, were reduced in this cohort. Our observed reduction in tonic seizures complements prior reports indicating steroids can reduce epileptic spasms, myoclonic seizures, and atonic seizures [10, 36, 37]. Additional EEG changes were seen in background organization and sleep. This suggests that video-EEG monitoring that captures wakefulness and sleep is important for assessments of corticosteroid efficacy. Repeated courses of steroids may be needed to reduce epileptiform burden [11].
Parental reports in this study support the importance of integrating assessments of cognition in corticosteroid clinical trials and studies of epileptic encephalopathies [11, 13]. Many other studies have identified cognitive and behavioral measures as important in epileptic encephalopathy outcomes [3]. For example, in one study examining diverse corticosteroid treatment among children with epilepsy, 21/36 (58%) of the treatment courses resulted in improved cognition or behavior [11]. However, typically used measures such as developmental quotient and full-scale IQ testing lack the sensitivity to see smaller changes in functional outcomes [38-40]. Sensitive and accessible measures for cognitive function in pediatric patients with severe neurologic disability are desperately needed.
Implications for epilepsy pathophysiology
The effect on tonic seizures and improvements in sleep spindles suggest corticosteroids affect thalamo-cortical or brainstem circuits [41, 42]. Animal models suggest that tonic seizures are produced from either a hyperexcitable response from thalamo-cortical input or abnormal brainstem functions. Corticosteroids may result in improved tonic inhibition of GABAA receptors within the thalamus and thereby reduce thalamocortical neuron excitation [43-45]. Examination of the effects of corticosteroids on generalized-onset motor seizures may provide insight into the pathophysiology of epileptic encephalopathies.
Limitations and identification of gaps for future investigation
The limitations of this study inform upon current gaps to be addressed in future studies. Two distinct and standardized steroid regiments were examined in this cohort; individuals receiving an altered steroid regimen were excluded, leading to possible selection bias. Patients who improved after a short course of steroids or patients who experienced significant side effects were likely excluded. Treatment with steroids earlier in the course of other epileptic encephalopathies, such as West syndrome, has been shown to be more effective [46], and therefore, true efficacy of corticosteroids may be underestimated by this study.
Use of EEG assessments is another domain to be improved upon in future study. The timing of EEGs in this cohort was heterogenous. Novel qualitative EEG measures were used. Though we found that inter-reader reliability was good, additional evidence with more readers and a more diverse set of EEGs would strengthen the validity of the measures. Clinical experts utilizing corticosteroids frequently have noted a reduction in epileptiform activity and improved ability to localize epileptic discharges which was not captured here. Future work might employ quantitative measures as a more sensitive measure of epileptiform activity compared to the qualitative scales used here. This study adds to other calls for reliable and sensitive EEG assessments for patients with coma, hypsarrhythmia and epileptic encephalopathies [47-51].
Clinical outcome assessments were also limited. Side effects here were based upon parental report and therefore may be under-reported. Additionally, these captured only short-term side effects; though long-term effects may be of concern. The cognitive improvement with corticosteroid treatment is based on parental report. Objective cognitive assessments are needed to validate these subjective reports. Like EEG assessments, there is a clear need for better outcome assessments in interventional trials that will incorporate standardized side effect profiling and cognitive and behavioral domains.
Last, given the small size of the cohort and the multiple comparisons post-hoc, we would interpret the statistically significant findings as hypothesis-generating.This adds to the need for larger, multicenter studies in the future that will validate these findings.
Conclusion
Future studies to examine the effects of corticosteroids will need to be large and capture patients earlier in the disease process, in order to clarify efficacy and maximize benefit of steroid use. Multi-institutional collaborative approaches may mitigate challenges posed by the heterogeneity of the epileptic encephalopathy population. Further work is needed to:
- –quantify and estimate cognitive improvement;
- –develop sensitive measures of EEG “spike burden”;
- –and further validate usable EEG assessment scales with good interrater reliability.
Supplementary data
Supplementary tables are available on the www.epilepticdisorders.com website.
Acknowledgements and disclosures
ZG received supported from the National Center for Advancing Translational Science of the National Institute of Health under award number UL1TR002384. ZG received research funding from the Pediatric Epilepsy Research Consortium, the BAND foundation, Weill Cornell Medicine, the Centers for Disease Control and Prevention, the Morris and Alma Schapiro Fund, the Children's Hospital of Philadelphia Orphan Disease Center, and Clara Inspired. JG was supported by the Neurological Sciences Academic Development Award, NIH-NINDS K12 award number NS066274-10.
None of the authors have any conflict of interest to declare.