Epileptic Disorders
MENUPossible critical region associated with late-onset spasms in 17p13.1-p13.2 microdeletion syndrome: a report of two new cases and review of the literature Volume 24, issue 3, June 2022

- Key words: 17p13.1–13.2 microdeletion, late-onset spasms, epileptic spasms, USP6, NMDA
- DOI : 10.1684/epd.2022.1416
- Page(s) : 567-71
- Published in: 2022
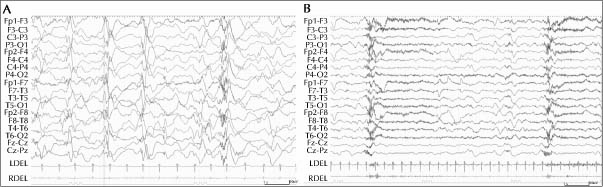
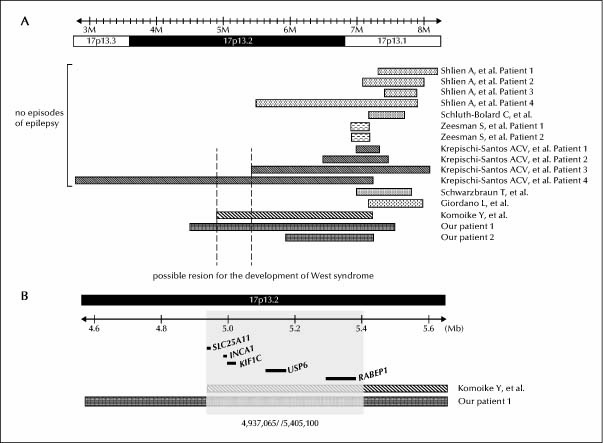
17p13.1-2 microdeletion syndrome is a congenital anomaly syndrome with characteristic facial features and multiple malformations. The prevalence of epilepsy with 17p13.1–2 microdeletion is low, with only one case reported for late-onset spasms. Late-onset spasms is one of the rare epilepsy syndromes and one of the developmental epileptic encephalopathies requiring urgent treatment. We experienced two cases of 17p13.1-2 microdeletion syndrome, one of which presented with epileptic spasms in cluster at 18 months of age. EEG showed symmetrical hypsarrhythmia during interictal periods and a paroxysmal fast wave superimposed on widespread slow waves during seizures, leading to the diagnosis of late-onset spasms. Another case had no epilepsy. Comparing the extent of deletion in the two cases with that of previous reports, the involvement of the USP6 gene was suspected. However, the accumulation of additional case reports is needed to confirm the genetic involvement in late-onset spasms.