Epileptic Disorders
MENUGenetic literacy series: genetic epilepsy with febrile seizures plus Volume 20, issue 4, August 2018

Figures
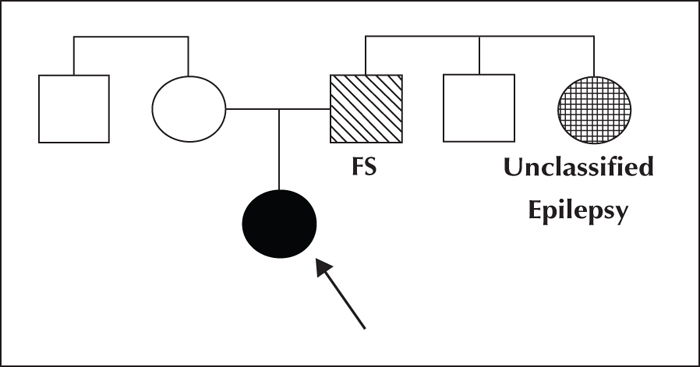
Illustrative case study
A three-year-old girl, accompanied by her mother, is seen in the emergency room for a new type of seizure. She had been having seizures every two to three months, since the age of 18 months. The seizures involved bilateral stiffening and clonic movements, with eyes deviated upwards, lasting 30 to 90 seconds. All had occurred with temperatures between 38.5oC and 40oC, when she was ill with rhinorrhoea and coughing. Her mother was told that these were febrile seizures (FS) and that the girl did not require any investigations or treatment. However, she now presents with another similar seizure, this time with a temperature of 36.8oC and no symptoms of illness.
The girl was born following an uncomplicated pregnancy and delivery at term. Her past medical history is otherwise unremarkable. There had been no concerns regarding her development; she sat at six months and walked independently at 12 months. She spoke her first word at one year, and is speaking in two to three-word sentences. Her general and neurological examinations were normal.
Based on the family history (figure 1), the girl's father had “convulsions” as a baby when he was sick, which disappeared as he grew up. The girl also has a paternal aunt with epilepsy; the girl's mother does not know the details of the aunt's presentation, but will ask the girl's father.
Questions
- (1)What is the girl's syndromic diagnosis?
- (2)What counselling is appropriate?
- (3)What, if any, genetic testing should be considered/offered?
GEFS+ spectrum
Genetic (formerly “generalized”) epilepsy with febrile seizures plus (GEFS+) was conceived as a familial epilepsy syndrome in which affected individuals within a family typically have a variety of epilepsy presentations or phenotypes (Scheffer and Berkovic, 1997). FS are the most common phenotype in the GEFS+ spectrum, followed by febrile seizures plus (FS+). Additional phenotypes in the GEFS+ spectrum include FS or FS+ with other generalized seizure types such as absence, myoclonic, atonic seizures or focal seizures (Abou-Khalil et al., 2001; Zhang et al., 2017). The majority of affected individuals have mild epilepsy with normal development.
The developmental and epileptic encephalopathies, Dravet syndrome (DS) and epilepsy with myoclonic-atonic seizures (MAE), also occur in GEFS+ families (Scheffer and Berkovic, 1997; Singh et al., 1999; Singh et al., 2001; Zhang et al., 2017). Within GEFS+ families, there may also be individuals with focal epilepsy or genetic generalized epilepsy phenotypes without FS/FS+ (Zhang et al., 2017). A GEFS+ family is defined as a family with ≥two individuals with GEFS+ phenotypes, including at least one with FS or FS+.
Key clinical features of the epilepsy syndromes within the GEFS+ spectrum are summarized as follows.
Febrile seizures (FS)
Febrile seizures are the most common phenotype in GEFS+, present in 41% of affected individuals in GEFS+ families (Zhang et al., 2017). FS are tonic-clonic seizures associated with fever (≥38oC), occurring between three months and six years (Consensus Development Panel, 1980; Scheffer and Berkovic, 1997). Seizures are typically bilateral convulsions and spontaneously terminate within several minutes in most cases, but may continue for more than 30 minutes (Chevrie and Aicardi, 1975; Storz et al., 2015; Vitaliti et al., 2017). FS are the most common seizure disorder with an estimated prevalence of 3-4%, though certain ethnic groups, including those from Finland, Japan and Guam, have an increased frequency (Van den Berg and Yerushalmy, 1969; Hauser and Kurland, 1975; Verity et al., 1985; Annegers et al., 1987; Hamdy et al., 2007). The precise percentage of patients with FS that fall within the GEFS+ spectrum is unclear. In one study, 6% of FS cases had a history of seizures, febrile or afebrile, affecting a first-degree family member (Nelson and Ellenberg, 1990).
Febrile seizures plus (FS+)
FS+ occur in 20% of affected individuals in GEFS+ families (Zhang et al., 2017). Of patients initially presenting with FS, 9% of cases had FS+ (Eckhaus et al., 2013). The FS+ syndromic diagnosis was initially defined as individuals with FS that continued past six years, with or without co-occurring afebrile generalized tonic-clonic seizures (Scheffer and Berkovic, 1997). The definition has subsequently been broadened to include two criteria, and patients can have one or both.
First, FS that extend beyond the typical age range (i.e. before three months or after six years). Second, the occurrence of both febrile and afebrile generalized tonic-clonic seizures, either limited to the usual age for FS (three months to six years) or occurring outside that time period (Eckhaus et al., 2013). Most commonly, generalized tonic-clonic seizures continue after age six years, with or without fever, and typically stop by adolescence, although rare adult afebrile generalized tonic-clonic seizures may occur (Singh et al., 1999; Singh et al., 2001). FS+ typically occur in individuals of normal intellect (Zhang et al., 2017).
Complex GEFS+ phenotypes
FS and FS+ can occur with a variety of other seizure types including absence, myoclonic or atonic seizures (Scheffer and Berkovic, 1997). Within GEFS+ families, focal seizures with temporal or frontal lobe semiology may also occur, not always preceded by FS (Abou-Khalil et al., 2001; Baulac et al., 2001; Scheffer et al., 2007). When FS or FS+ occur with other seizure types, this can be conceptualized as “FS or FS+ with _____ seizures” (e.g. “FS+ with absence seizures”).
In addition, classic genetic generalized epilepsies occur in 5% of affected individuals in GEFS+ families, sometimes with preceding FS (Zhang et al., 2017). Recently, afebrile generalized tonic-clonic seizures alone, without preceding FS, have been observed in GEFS+ families (Zhang et al., 2017). When these focal or generalized epilepsy syndromes occur without FS, but in the context of a GEFS+ family, they may also be considered to lie within the GEFS+ spectrum.
Associated developmental and epileptic encephalopathies
GEFS+ may involve more severe epilepsy syndromes; specifically, DS and MAE (Scheffer and Berkovic, 1997; Singh et al., 1999; Singh et al., 2001). DS is a developmental and epileptic encephalopathy that classically presents with FS at around six months, often with hemiclonic status epilepticus. Other seizure types subsequently develop, most commonly including afebrile generalized tonic-clonic, focal impaired awareness, myoclonic, and absence seizures (Dravet, 2011). Development is initially normal, however, by two years of age individuals usually show developmental plateauing or regression, and ultimately have intellectual disability that varies from severe to mild.
MAE (formerly known as myoclonic-astatic epilepsy or Doose syndrome) is a childhood-onset generalized epilepsy characterized by drop attacks which are myoclonic-atonic, myoclonic or atonic in nature, with onset between two and six years of age. Patients typically develop additional seizure types including absence and generalized tonic-clonic seizures, as well as episodes of non-convulsive status epilepticus; focal seizures are not seen. In two thirds of cases, FS precede the myoclonic-atonic and atonic seizures (Bureau et al., 2012). Early development is typically normal with regression at seizure onset, indicating that it should be considered an epileptic encephalopathy in at least some cases. Long-term developmental outcome is variable; some individuals have normal intellect while others have intellectual disability (Bureau et al., 2012).
Genetic architecture of GEFS+
Inheritance
GEFS+ was originally described in a large Australian family, in which inheritance followed an autosomal dominant pattern with variable expressivity (Scheffer and Berkovic, 1997). Subsequently, many families have been reported with a similar pattern of inheritance (Baulac et al., 1999; Singh et al., 1999), leading to the initial concept that GEFS+ was an autosomal dominant disorder.
However, in smaller families and sporadic cases, inheritance may be polygenic or due to de novo mutations (Wallace et al., 1998; Singh et al., 1999; Eckhaus et al., 2013; Myers et al., 2017). Further, two small consanguineous GEFS+ families have been reported with likely autosomal recessive inheritance (Brunklaus et al., 2015). As GEFS+ phenotypes have become better recognized, it is clear that not all occur in a familial context, and they can be diagnosed in sporadic cases, in which de novo mutations in GEFS+ genes may be found (Myers et al., 2017).
Implicated genes
SCN1A
SCN1A (OMIM #182389) encodes the alpha-1 neuronal voltage-gated sodium channel subunit Nav1.1, and was the second gene associated with GEFS+ to be discovered (Escayg et al., 2000). Of GEFS+ patients with known mutations, SCN1A mutation accounts for the largest fraction, with mutations identified in 19% of families (Marini et al., 2007; Zhang et al., 2017). Although inheritance is usually autosomal dominant with variable expressivity and penetrance, de novo mutations and autosomal recessive inheritance have also been described (Brunklaus et al., 2015; Myers et al., 2017). Patients with FS/FS+ due to SCN1A mutations have an earlier median onset of FS of 11-12 months compared with the population median of 18 months (Sijben et al., 2009).
SCN1A mutations are identified in >80% of DS patients, and the vast majority of individuals have de novo mutations. However, for approximately 10% of apparently “de novo” inheritance, one parent is a mosaic carrier and, depending on the percentage mosaicism, may exhibit a milder phenotype including FS or FS+ (Xu et al., 2015). GEFS+ is typically associated with missense SCN1A mutations, while DS may occur with both missense and truncation mutations (Zuberi et al., 2011).
SCN1B
SCN1B (OMIM #600235) encodes the beta-1 subunit of the voltage-gated sodium channel. SCN1B was the first gene discovered for GEFS+, described in a large family with autosomal dominant inheritance with variable expressivity (Wallace et al., 1998). SCN1B mutations are identified in up to 8% of GEFS+ families (Zhang et al., 2017). Patients with FS/FS+ and SCN1B mutations have later onset of FS compared to patients with mutations in SCN1A (24 months versus 12 months) (Sijben et al., 2009). When focal epilepsy occurs in such GEFS+ families with SCN1B mutations, the phenotype is that of temporal lobe epilepsy which may or may not be preceded by FS (Scheffer et al., 2007).
GABRG2
Multiple GEFS+ families have been reported with mutations in GABRG2 (OMIM #137164), the gene encoding the GABA-A gamma-2 receptor subunit (Baulac et al., 2001; Wallace et al., 2001; Tian et al., 2013; Ishii et al., 2014). Overall, GABRG2 mutations are identified in up to 9% of GEFS+ families (Zhang et al., 2017). Individuals with genetic generalized epilepsies, particularly childhood absence epilepsy, may be more common in GEFS+ families with GABRG2 mutations (Wallace et al., 2001; Marini et al., 2003).
Other genes
Other genes have been implicated in GEFS+ based on the identification of mutations in families or individuals with FS+, including STX1B (Schubert et al., 2014), SCN9A (Singh et al., 2009; Mulley et al., 2013), GABRD (Dibbens et al., 2004), and FGF13, and have been suggested as susceptibility alleles for GEFS+ (Puranam et al., 2015; Rigbye et al., 2016), however, an understanding of the role of these genes requires further investigation.
Variants in SCN2A have also been reported in a small GEFS+ family (Sugawara et al., 2001) and a single individual with FS and childhood absence epilepsy (Haug et al., 2001). However, both variants have subsequently been identified in control populations suggesting that they may not be pathogenic or perhaps contribute as a susceptibility allele for the phenotype in these individuals.
Current practical management considerations
When faced with a patient such as the girl presented here, making a correct diagnosis with appropriate counselling and judicious use of genetic testing is essential. This girl had febrile and afebrile generalized tonic-clonic seizures, thus her diagnosis is FS+.
The first step is discussing the epilepsy diagnosis with the family, including the likelihood of seizure recurrence, need for medical therapy, and risk of co-morbidities, including the risk of sudden unexpected death in epilepsy. The family can be counselled and informed that for most individuals with FS+, seizures will spontaneously resolve by puberty, however, there is a small risk of seizures occurring in adult life. Development is expected to be normal.
Families may ask whether evolution to DS is possible; in the present case, the patient's presentation is clearly not in keeping with DS. However, when infants present in the first year of life with febrile and afebrile convulsions, differentiating DS from FS+ may not be straightforward; we suggest following with developmental surveillance, serial EEGs, and counselling with a “time will tell” approach until there are clinical features that allow for more clear differentiation.
Regarding the risk for future offspring, inheritance in the small family of the presented case appears to be autosomal dominant with variable penetrance, though other modes of inheritance are possible. The parents of the girl can be informed that the risk for any of their future offspring is likely to be less than 50% based on the available information.
Regarding genetic testing, there is ongoing debate as to whether this is appropriate for individuals with GEFS+ phenotypes (Hirose et al., 2013; Myers et al., 2017; Zhang et al., 2017). Some argue that, while clinical care is unlikely to change, identifying a mutation would be useful in terms of prognosis and family planning. If genetic testing is offered, SCN1A sequencing should be included, although this would be more efficiently performed as part of a panel or via whole-exome sequencing. In the presented case, the family should be informed that although the girl and her father (with FS) both appear to be mildly affected, there is a potential for other individuals who inherit a mutation affecting certain genes to have a more severe phenotype.
Resolution
In the present case, the girl was diagnosed with GEFS+. Her parents elected to undergo whole-exome sequencing which did not reveal any mutations in known GEFS+ genes. The parents subsequently had a second child, who was healthy and developmentally normal, and did not have seizures. The first girl was treated with valproate and was seizure-free for two years, at which point she was weaned off medication. She was last seen at six years of age, at which time she had been seizure-free off medication for over a year. She was discharged from the clinic, however, the family was informed that she has an ongoing elevated risk for recurrent seizures, in which case she should return to the clinic.
Supplementary data
Summary didactic slides are available on the www.epilepticdisorders.com website.
Disclosures
IES and SFB are named inventors on a patent for diagnosis involving SCN1A. The patent, held by Bionomics Inc., is licensed to Athena Diagnostics and Genetics Technologies Ltd. KAM has no conflict of interest to declare.
This report was written by experts selected by the International League Against Epilepsy (ILAE) and was approved for publication by the ILAE. Opinions expressed by the authors, however, do not necessarily represent the policy or position of the ILAE.
Key points
- –The most common GEFS+ phenotype is febrile seizures (FS) followed by febrile seizures plus (FS+).
- –FS+ is defined as either or both of the following: (A) febrile tonic-clonic seizures that begin before three months and/or continue after six years; (B) the occurrence of both febrile and afebrile generalized tonic-clonic seizures.
- –The GEFS+ spectrum comprises a heterogeneous group of generalized and focal epilepsy phenotypes including the developmental and epileptic encephalopathies of epilepsy with myoclonic-atonic seizures and Dravet syndrome.
- –Mutations in SCN1A are the most common identified genetic cause of GEFS+, accounting for 19% of families.
- –Whether clinical molecular genetic testing in patients with FS+ or other GEFS+ phenotypes is indicated is currently under debate. If genetic testing is offered, SCN1A sequencing should be included, and this may be most efficiently performed as part of a panel or whole-exome sequencing.