Epileptic Disorders
MENUEpigenetics explained: a topic “primer” for the epilepsy community by the ILAE Genetics/Epigenetics Task Force Volume 22, issue 2, April 2020

Epigenetics is a word that has, within just a few years, become common to hear at epilepsy conferences. But what does it really refer to? In 2017, the Neurobiology Commission of the International League Against Epilepsy (ILAE) established a new Task Force that was given the specific remit of exploring the topic, how it intersects with ongoing genetic discoveries about causes of epilepsy and whether it will lead to new therapeutic approaches. Why a “primer” on epigenetics? We are in the midst of an exponential phase of discovery about how gene expression is controlled. This has been driven, in part, by advances in the field of cell biology and gene sequencing technology. Together, these allow researchers to ask questions about the molecular mechanisms which shape the gene expression landscape. This not only helps our understanding of the neurobiology of disease but offers opportunities to discover biomarkers and new treatments for patients. Researchers in the areas of neurodevelopment and psychiatry have been first to embrace and apply these technologies to the brain. Practical applications may soon reach the clinic, including therapies that target epigenetic processes, molecular diagnostics and gene panels. We, in the epilepsy field, risk falling behind and must catch up, fast.
The Genetics-Epigenetics Task Force recognised a first step was to provide an overview of the subject and its potential practical applications. Our target audience are clinical colleagues and basic scientists interested in the mechanisms and treatment of epilepsy. We select examples of work from within the field and outside, including studies by expert members of the Task Force. The result, we hope, is a clear and understandable set of definitions and explanations of the main mechanisms at work. We include a glossary of terms (Box 1) and Frequently Asked Questions (FAQs, Box 2).

We review some of the key findings, provide evidence that epigenetic processes underlie epileptogenesis and the epileptic state, and look at prospects for epigenetic-based therapies and biomarkers. Finally, we discuss the limitations of the work to date, particularly around translation to clinical practice, and some of the next questions that should be asked and, hopefully, answered (Box 3).
The manuscript is addressing between others, issues corresponding to the knowledge domain 7.0 (Biology of Epilepsy) and competency 1.2 (related to understanding genetic testing procedures) of the ILAE curriculum (Blümcke et al., 2019).
What is epigenetics?
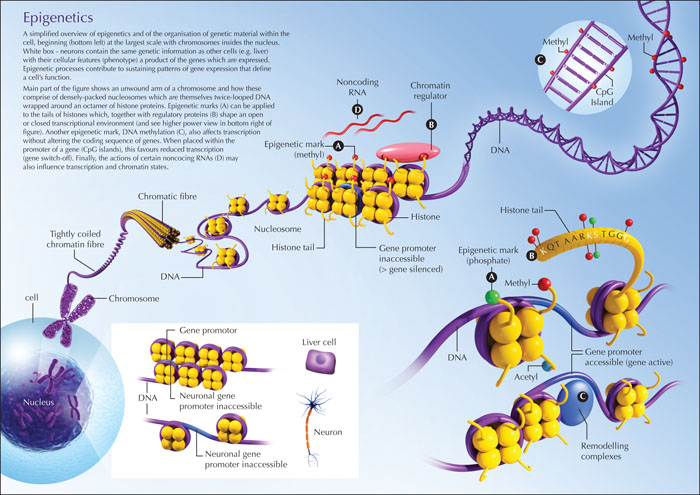
Before tackling epigenetics, let us briefly step back and ask what makes a neuron a neuron? The functional properties of a cell are generated by the genes that are expressed. Just because you have a gene that codes for something (say an ion channel) does not mean a particular cell will make the protein for which it encodes. The brain transcribes about 80% of all the genes in our genome (Kang et al., 2011; Hawrylycz et al., 2012). Some genes are transcribed by all cells regardless of specialized function while others are not. For example, neurons express various channel proteins that allow the generation and propagation of action potentials. An efficient way to tackle this is for genes that are never needed to be more or less permanently switched off while those needed often are kept in a ready-to-go state. To produce a protein, transcription factors must bind to a promoter region of a gene and the RNA (ribonucleic acid) polymerase machinery must assemble, generating a messenger RNA that, following a series of further processing and trafficking events, will be translated into a protein. That first step – getting access and reading the code for transcription – is where epigenetics occurs. Epigenetics (Box 1) is the umbrella term for the collection of biochemical processes which influence the readability of the genetic code. Put another way, epigenetic processes determine whether or not a gene can be “read”. This is achieved by controlling the structure of chromatin – the packaged DNA inside the nucleus – which in turn dictates accessibility of the genetic code to transcription. Epigenetics works, primarily, through adding or removing chemical tags on either DNA or on the proteins around which DNA is wrapped (histones). The nucleus of our cells possess an extensive assortment of proteins and RNAs which work together to apply, read and wipe off these marks. These epigenetic marks cause physical changes to chromatin, making it more open (termed euchromatin) or less open (heterochromatin) which makes transcription easier or harder, respectively. Together, this creates an appropriately stable but still responsive gene expression environment in the cell. While epigenetic marks are heritable and can be passed on during cell division, many of the underlying mechanisms are functional and responsive to stimuli in post-mitotic cells, including neurons.
The complexity of epigenetics is staggering. For example, it is estimated that there are ∼600 potential modifications to histones that can affect transcription. While we have a very good understanding of some epigenetic processes such as the inactivation of the spare X chromosome in female cells, we do not have answers to how most other individual or collective epigenetic marks shape gene expression in health and disease. This lack of knowledge is slowly being resolved and it is possible that a fairly complete “epigenome” will be possible to map for most diseases, including the epilepsies. There is also a strong age-dependent aspect to epigenetics, with chromatin becoming increasingly “closed” as we age, including within the brain (Berson et al., 2018). The implications of this are currently unknown. For the purposes of our Task Force, we will use the following definition of the epigenome, based on that of the NIH: “The chemical modifications added to the entirety of one's DNA (genome) as a way to regulate the activity (expression) of all the genes.”
The basic epigenetic processes
There are three main categories of biochemical mediators of the epigenetic control of gene expression. All are important in brain development, function and disease. These are:
- –methylation of DNA;
- –histone modifications and variants;
- –and non-coding RNAs.
Each comprises a varying degree of complexity in terms of the mediators involved and their influence on gene transcription and are facilitated by a further set of proteins that form chromatin remodelling complexes (Dulac, 2010; Jakovcevski and Akbarian, 2012).
DNA methylation
DNA methylation refers to the addition of a methyl (-CH3) group that occurs on cytosine bases (at the carbon 5 position [5mC]) in the genetic code (figure 1). This may occur at various sites within and outside the coding region of a gene. It can occur in regions that contain promoter elements for a gene, but can also occur at exon/intron boundaries and intergenic (between genes) regions. Some regions of the genome are particularly densely methylated, such as those encoding transposons (DNA sequences that can change their position within a genome). The primary biological functions of DNA methylation includes maintenance of genomic stability by silencing transposable elements (including retrotransposons), the monoallelic expression of imprinted genes, X chromosome inactivation in female cells, and the selective exposure of promoters and enhancers of genes to transcription factors (Edwards et al., 2017; Greenberg and Bourc’his, 2019). A number of genes have been identified that undergo changes in methylation in response to synaptic activity. Thus, DNA methylation marks support the gene expression programmes that underlie synaptic plasticity, producing lasting and responsive adjustment to the expression of certain genes (Sweatt, 2013). DNA methylation is mediated by DNA methyltransferases (DNMT1, 3A and 3B). The process is reversible through base excision repair proteins (Edwards et al., 2017).
Histone post-translational modification and variants
Packaging the approximately six billion bases of DNA within our cell's nucleus is a major challenge. It is achieved by winding DNA around a set of proteins called histones and then packing individual units, termed nucleosomes, together to eventually form a chromosome (figure 1). Importantly, densely packed nucleosomes are inaccessible to the machinery of transcription whereas loosely-packed chromatin can be read (i.e. genes transcribed). The nucleosome comprises a 146 base-pair stretch of DNA around an octamer of histone proteins; two copies each of H2A, H2B, H3 and H4. The amino terminals (tails) of these histones protrude from the nucleosome and, along with the core region, are subject to modifications, including acetylation, phosphorylation, methylation and ubiquitylation. These biochemical additions lead to changes in chromatin compaction, in part, by altering charges on the proteins so that they move closer or further apart. The influence of these modifications is complex and context-specific and predicting their effects is challenging. The transcriptional landscape cannot [yet] be determined by reading the overall histone code. However, we know that increased acetylation of histones is associated with increased transcription. This is mediated by acetyl transferases and the mark is removed by histone de-acetylases (HDACs; thus, a histone deacetylase inhibitor would tend to promote gene expression; see below). Other histone marks are associated with reduced gene expression, such as methylation of lysine residues on H3 (H3K9me2, H3K27me3). In addition to post-translational modifications, variants of the histones can replace the canonical histones in the nucleosome. This promotes unique interactions with other factors that shape chromatin structure (Berson et al., 2018). From a research perspective, it is possible to study either individual histone changes or globally assess histone changes, for example, by breaking up modified histones and the segments of DNA to which they are localised.
Non-coding RNAs
The third major category of the epigenetic process is the role of non-coding RNAs. This is a highly heterogeneous class of RNAs that are transcribed but do not code for proteins (unlike messenger RNA). They are split into short (or small) non-coding RNAs (
The non-coding RNAs that mediate epigenetic effects are considered to belong to the main classes of short and long non-coding RNAs (based on a length of 200 bases). Long non-coding RNAs have been identified which, upon transcription, serve as structural bridges to bring together segments of chromatin called “loops” (St Laurent et al., 2015). The bringing together of such loops can be an important influence on gene transcription. Long non-coding RNAs can also cause changes to histone modifications, directly influence DNA methylation, and alter other aspects of chromatin structure (Hanly et al., 2018). A number of long non-coding RNAs have been identified with roles in brain function, influencing synaptic structure and neurophysiological properties of neurons (Bernard et al., 2010; Barry et al., 2017). Over the last two decades, several types of small non-coding RNAs with epigenetic functions have been discovered. These include microRNA (miRNA), piwi-interacting RNA (piRNA), enchancer RNA (eRNA) and natural antisense transcripts (NAT). The most widely studied class of short RNA are miRNAs. These mainly function outside the nucleus, binding to mRNA to reduce protein production (i.e. acting post-transcriptionally). They generally do not directly perform epigenetic functions. Aberrant expression of miRNAs can, however, lead to alterations in the proteins that serve epigenetic functions, for example by restricting the activity of chromatin re-modelling enzymes (Wei et al., 2017). Indeed, miRNAs are capable of targeting genes that control epigenetic pathways, including histone methyltransferases, methyl CpG binding proteins and histone deacetylases (Lewis et al., 2005; Sato et al., 2011).
Among other small non-coding RNAs, piRNAs have been reported to be involved in epigenetic regulation by modulating histone modifications and DNA methylation (Esteller, 2011; Luteijn and Ketting, 2013). Enhancer RNAs (eRNA) are a class of short non-coding RNA that act within the nucleus to promote or oppose transcription of specific genes. They possess some classic epigenetic characteristics since they can modify histones, may contribute to a more open chromatin structure and help bring enhancers and promoters together through chromatin looping (Ding et al., 2018). NATs are transcribed from the opposite strand of DNA from the side that encodes a gene and compete locally to reduce abundance of the coding mRNA (Wahlestedt, 2013). Both long and short non-coding RNAs have been explored for therapeutic potential in epilepsy (see below).
Other mediators of epigenetic processes
In addition to the three main processes described in the previous paragraphs, a number of additional mediators are involved in epigenetic functions. These include the chromatin remodelling factors that are recruited to a genomic location after a gene promoter is methylated, which perform the subsequent mechanical aspects of condensing chromatin. Transcription factors – both activators and repressors – often work in tandem with epigenetic factors. For example, neuron restrictive silencing factor (NRSF) is attracted to sites with specific sequences whereupon it attracts histone deacetylases to mediate gene silencing (Huang et al., 1999).
Epigenetic mechanisms in epilepsy - neurobiological implications
All three major epigenetic processes have been observed, and found altered, in experimental and human epilepsy (Henshall and Kobow, 2015). This includes changes to DNA methylation, histone post-translational modifications, and changes to the expression or activity of non-coding RNAs. Researchers have performed both focused work, looking at how epigenetic marks at specific loci affect gene expression as well as several studies that have attempted to characterise the totality of an epigenetic mark across the entire genome. Collectively, studies about epigenetic changes in epilepsy indicate that epigenetic processes serve some role in shaping the gene expression landscape during epileptogenesis and in chronic epilepsy. However, our understanding of the mechanisms involved in applying, reading and removing epigenetic marks is poor and we do not yet have the ability to control these or predict their effects which will be essential for any epigenetic-based treatment for epilepsy.
DNA methylation in epilepsy
The DNA methylation landscape has been mapped in both experimental and human epilepsy. The first genome-wide study of DNA methylation in human epilepsy determined that most DNA methylation was unchanged in hippocampal samples from patients with temporal lobe epilepsy (TLE) compared to control brain (Miller-Delaney et al., 2015). Select increases (hyper-) and decreases (hypo-) in methylation were found but few of these occurred around genes that had conceivable links to pathomechanisms of epilepsy. Moreover, altered DNA methylation was often not predictive of associated changes in levels of gene transcription, meaning other factors were important (Miller-Delaney et al., 2015). Important caveats in this study were small sample size and technical bias introduced when comparing surgically-obtained material to autopsy control. Kobow et al. performed the first genome-wide analysis of DNA methylation in chronic epileptic rats and identified epilepsy-associated genome-wide DNA methylation changes which correlated well with gene expression. That is, genes that were highly methylated showed a reduction in expression and vice versa (Kobow et al., 2013). In a follow up study, Debski et al. found genome-wide changes in DNA methylation to be a common feature of several different pro-epileptogenic insults. These epigenetic changes were unlikely to be an epiphenomenon as they appeared highly specific with distinct aetiology-dependent DNA methylation patterns observed across different acquired epilepsy models (i.e. pilocarpine, amygdala stimulation, and traumatic brain injury [Debski et al., 2016]). DNA methylation profiling may, therefore, be clinically useful to diagnose and stratify epilepsy patients. This was recently tested and validated in epilepsy patients with different focal cortical dysplasia (FCD) subtypes, TLE patients and non-epilepsy controls (Kobow et al., 2019). Where there are discrepancies between changes in methylation and gene expression, a possible explanation may derive from competing epigenetic mechanisms besides DNA methylation. Further research is required to better understand what drives these changes in DNA methylation marks and how they influence, or not, gene expression in epilepsy.
Technological advances are transforming capabilities to map the gene expression landscape. Among key recent advances is the ability to study the 3D genomic landscape using techniques such as ATAC-Seq (Assay for Transposase-Accessible Chromatin Sequencing) (Buenrostro et al., 2013). This allows mapping of all open and closed chromatin sites in the genome, significantly improving our understanding of mammalian gene regulation in normal development and in pathological conditions (Bonev et al., 2017). These techniques provide an exciting opportunity to extend our understanding of gene to genome interaction and gene regulation in epilepsy.
Histones in epilepsy
A number of studies have reported post-translational histone modifications in epilepsy. The earliest work reported acetylation changes to histones located at the loci of glutamate receptors and brain derived neurotrophic factor (Huang et al., 2002). These studies revealed that histone modulation influences transcription of genes that regulate brain excitability. Other studies have also identified phosphorylation of histones within the hippocampus after experimental status epilepticus (Crosio et al., 2003; Sng et al., 2006). There are now methods that can globally assess histone modifications. These typically involve fragmenting chromatin and using an antibody specific to a certain histone modification to pull out regions followed by sequencing. There has not yet been a global characterisation of histone modifications in experimental or human epilepsy.
Non-coding RNAs in epilepsy
Both long and short non-coding RNAs have been reported to be dysregulated in experimental and human epilepsy. There is evidence linking several long non-coding RNAs to the control of neuronal excitability, including BC1 (Zhong et al., 2009), MALAT1 (Bernard et al., 2010) and NEAT1 (Barry et al., 2017). Profiling studies using gene arrays and RNA sequencing have revealed the dysregulation of numerous long non-coding RNAs (Lee et al., 2015; Miller-Delaney et al., 2015; Jang et al., 2018; Xiao et al., 2018) and short non-coding RNAs (Henshall et al., 2016; Korotkov et al., 2017; Srivastava et al., 2017; Cava et al., 2018; Mills et al., 2019) in experimental and human epilepsy. However, few functions have been assigned to these transcripts. The only class of short non-coding RNA for which we have extensive evidence of roles in epilepsy is miRNA (Henshall et al., 2016). Some miRNAs that function in an “epigenetic way” have been identified, including miR-320 (Kim et al., 2008; Catalanotto et al., 2016). However, these have not been manipulated in experimental models to date, making it difficult to assign a biological role. Moreover, studies that have tested miRNA manipulations in seizure models have not investigated whether this produces changes to the epigenetic landscape, an area that could be explored in the future. Intriguingly, a number of miRNAs are mis-localised to the nucleus in the hippocampus of patients with drug-resistant temporal lobe epilepsy (Kan et al., 2012; Raoof et al., 2018). This could indicate that miRNAs acquire new (epigenetic) functions in epilepsy. Further research is needed to explore the expression and function of other non-coding RNAs including eRNA which serves more traditional epigenetic roles.
Epilepsy genes with epigenetic functions
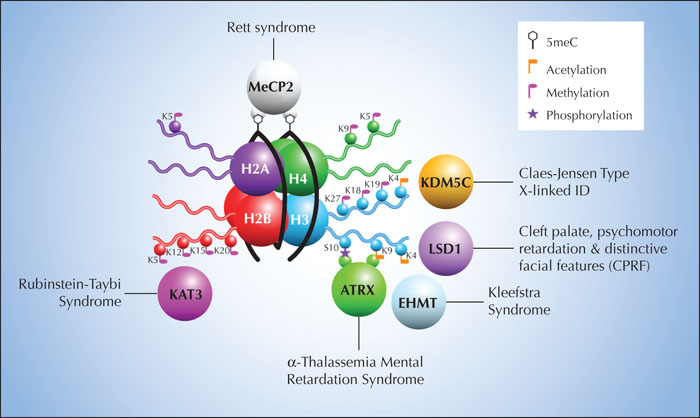
Mutations in a growing number of genes which are neither ion channels, receptors nor other classic epilepsy genes, but instead encode proteins that serve epigenetic functions, have been identified as responsible for genetic forms of epilepsy (table 1, figure 2). The list of clinical phenotypes includes epileptic encephalopathies, intellectual disability syndromes and autism spectrum disorders with associated severe or occasional seizure phenotypes. A good example is CHD2, mutations in which cause a Dravet-like fever associated epileptic encephalopathy (Suls et al., 2013). This gene codes for an ATP-dependent helicase with chromatin remodelling function (Carvill et al., 2013; Suls et al., 2013). Re-modellers such as the CHD protein family alter gene expression by modification of chromatin structure, meaning that they can help to improve accessibility of the transcriptional apparatus to the DNA template either by moving, ejecting or restructuring nucleosomes. In an energy-dependent process, the remodelling complexes reposition (i.e. slide, twist or loop) nucleosomes along the DNA, expel histones away from DNA or facilitate exchange of histone variants, and thus create regions of DNA for gene activation (Marfella and Imbalzano, 2007). ATRX, that encodes another chromatin re-modeller, was found to be mutated in Alpha-thalassemia/mental retardation (ATRX), which is associated with seizures in about one third of patients (Gibbons, 2006). Mutations in SMARCA4, ATRX and other re-modeller genes have also been found to cause Coffin-Siris syndrome, with structural brain abnormalities and seizures as typical features (Tsurusaki et al., 2012).
Histone modifying enzymes and readers of histone tags are frequently identified in intellectual disability syndromes with associated epilepsy. The protein encoded by EHMT1 is a histone methyltransferase that is implicated in learning and memory, and was recently shown to be mutated in Kleefstra syndrome (Iwase et al., 2017). KDM5 encodes a histone demethylase, and as such functions to erase methylation marks from histones. Its function in the brain is not fully determined, but a mutation in this gene has been associated with X-linked intellectual disability (Poeta et al., 2013). Insertion/missense mutations of the transcription factor ARX have been linked to epileptic encephalopathy, lissencephaly and X-linked intellectual disability as well as functions including regulation of histone methylation patterns.
Mutations in genes that regulate DNA methylation have also been linked to epilepsy. MECP2 encodes a typical reader of DNA methylation that when mutated is the cause of classic Rett syndrome (Amir et al., 1999). Seizures are reported in up to 90% of affected females, with generalized tonic-clonic seizures and partial complex seizures being most common. Notably, MECP2 duplication syndrome is commonly associated with an epileptic encephalopathy and intellectual disability (Marafi et al., 2019). This indicates an intolerance of both lower and higher amounts of certain epigenetic factors. MBD5 encodes another DNA methylation binding protein and was identified as causing intellectual disability and epilepsy in patients with 2q23.1 microdeletion syndrome. Variation in CDKL5 that codes for a cyclin-dependent kinase has been associated with an X-linked dominant early infantile epileptic encephalopathy (Weaving et al., 2004). The CDKL5protein has been proposed to interact with MECP2 and DNMT1, thereby influencing gene expression and DNA methylation (Carouge et al., 2010).
A number of studies have reported variants in non-coding RNAs in epilepsy cases, including miRNAs (Manna et al., 2013; Cui et al., 2015; Manna et al., 2016), but these have yet to be replicated. Figure 2 provides an example of how multiple genes associated with epilepsy may be linked through epigenetic processes. It may be difficult to anticipate how a mutation affecting an epigenetic enzyme or modulator may influence seizure development. Studying the 3D genome in e.g. mice or cell lines with an epigenetic mutation may help to unravel long-ranging effects of these mutations (Barutcu et al., 2016) and better explain the connection to altered brain functions.
Epigenetic cross-talk in epilepsy
While the three major categories of epigenetic process are presented separately, they should not be thought of as functioning in silos. The main epigenetic processes do not act in isolation but instead interact and converge. As mentioned earlier, histone modifications can attract or expel components that regulate DNA methylation. As a result, assessing single epigenetic marks is often not predictive of overall gene expression when, in practice, each mark is likely to influence others. Such “collective” approaches to assessing epigenetic influences will be needed in the epilepsy field but have not been undertaken. One or two examples of over-lapping epigenetic mechanisms have been reported, including the observation that a subset of differentially expressed miRNAs and long non-coding RNAs were influenced by DNA methylation in epilepsy patients with hippocampal sclerosis (Miller-Delaney et al., 2015).
In which cells do epigenetic changes occur?
Studying epigenetic processes in vitro enables researchers to be confident of the cell types involved because relatively pure cell populations can be studied. This has confirmed roles for epigenetics in neuronal function and plasticity. However, all of the in vivo data we have for epilepsy, including genome-wide analyses of DNA methylation, is based on bulk tissue. We therefore do not know the cell source of the “signal” – it could be neurons (but which types?) or glia or other cells. The machinery necessary for epigenetic processes is not equally divided among cell types. For example, expression of DNMT1 and H3A is highly enriched in neurons and hardly detectable in glia (see www.brain-map.org). In theory, neurons and glia do not have parity in terms of how much the transcription of their genes is influenced by epigenetic processes – both adding and removing epigenetic marks. This will be important to resolve in the future, and the technology to separate individual neurons from bulk tissue and assay their epigenome will provide a means to determine where the epigenetic changes are really happening.
How do epigenetic changes lead to neuronal excitability and seizures?
Fundamentally, changes in neuronal network behaviour on a millisecond timeframe are responsible for the generation of seizures. However, the process by which a neuronal network becomes more susceptible to generating seizures can occur across a broad temporal scale. Furthermore, epigenetic changes by their nature have the potential to have far-reaching consequences on cellular function. Acute changes caused by epigenetic factors could include altered voltage and/or ligand-gated ion channel expression that would alter a neuron's moment-to-moment excitability. Epigenetic changes could also initiate cell death signalling (e.g. apoptotic) cascades over longer periods that result in neurodegeneration and imbalances in network excitability. An additional raft of consequences could be caused by epigenetic insults occurring early in development, including changes in axon guidance, synaptogenesis and neuronal migration, differentiation and proliferation leading to long-term changes in network stability. Attributing a causative action of a particular epigenetic-mediated change on neuronal function is therefore challenging. Syndromes in which causes have been isolated to a single “epigenetic gene” (table 1) are likely to have a narrower spectrum of cellular consequences. Genetically-modified mice for the study of “epigenetic genes” are available and are beginning to provide some insight into cellular mechanisms of excitability. As always, we do not know the extent to which animal model findings will translate to human epilepsy. Interestingly, physiological studies have indicated that cognitive deficits in murine models based on disease-causing epigenetic genes can be reversed in adulthood (Guy et al., 2007). This could have important treatment implications suggesting that a more dynamic role of epigenetic changes is responsible for at least some of the pathological outcomes in neurodevelopmental disease. In particular, studies have broadly implicated altered synaptic plasticity in neurodevelopmental disease caused by mutations of epigenetic genes (e.g. MECP2) (Johnston et al., 2015; Iwase et al., 2017). This understanding has motivated clinical studies aimed at targeting the pathomechanisms. For example, there is a marked increase in expression of the N-methyl-D-aspartate (NMDA) receptor, a glutamate receptor, in MeCP2 knock-out mice (Blue et al., 2011) that recapitulates clinical findings in Rett patients (Blue et al., 1999). Dextromethorphan, an NMDA antagonist, has been shown to improve speech in Rett patients, but did not reduce seizure liability (Smith-Hicks et al., 2017). Clearly, we are some way off fully elucidating the mechanisms that cause increased excitability in “epigenetic disease”. There may well be different mechanisms causing distinct clinical phenotypes (cognition vs seizures). The therapeutic time window in which treatment needs to be given will differ for different epigenetic insults. Converging pathomechanisms (e.g. synaptic plasticity) in some epigenetic diseases suggest that common treatment strategies may be possible.
Diagnostic implications
There are a number of potential diagnostic applications of the discoveries on epigenetic marks in epilepsy. Rodent studies clearly show that DNA methylation patterns differ between aetiologies (Debski et al., 2016). This could have practical implications in tissue diagnostics. Resected tissue has been reported to contain unique epigenetic signatures that could form the basis of classification alongside traditional histological findings (Miller-Delaney et al., 2015; Kobow et al., 2019). Epigenetic marks and factors are also detectable in circulating biofluids. This has been best explored for miRNAs which, due to their tissue-specific expression, stability and amenity to assay, have been studied the most (Enright et al., 2018). A number of miRNAs are found at altered levels in human epilepsy, including in plasma and cerebrospinal fluid, and research suggests the levels of these molecules change in response to therapies (Trelinska et al., 2016; Raoof et al., 2018). However, the analysis of circulating miRNAs is challenging, which relates both to the lack of standardized sampling protocols and to analysis of miRNAs. Recently, a protocol for standardization of procedures for discovery of circulating miRNA biomarkers in preclinical models has been proposed (van Vliet et al., 2017). This may facilitate clinical biomarker discovery. The technology to detect miRNAs in a point-of-care setting and commercial opportunities for miRNA-based diagnostics is being explored. Other epigenetic marks may also have diagnostic value. Distinct DNA methylation patterns in blood samples have been reported in patients with epilepsy compared to controls (Long et al., 2017). Again, the relative stability of DNA methylation would seem an ideal attribute for a biomarker.
Therapeutic implications and limitations
The discovery, in 2001, that sodium valproate had HDAC inhibitor activity (Gottlicher et al., 2001) drove excitement that this commonly used treatment for epilepsy may work through an epigenetic mechanism. However, other HDAC inhibitors lacked this property (Hoffmann et al., 2008). Nevertheless, there continues to be significant interest in using epigenetic drugs to treat brain diseases, including epilepsy. A number of anti-cancer therapies have been approved which have epigenetic mechanisms of action, including other HDAC inhibitors and inhibitors of DNA methylation. Whether any of these drugs could be re-purposed for use in epilepsy is unknown but could be the focus of future research.
A number of epigenetic manipulations have shown promising effects in experimental epilepsy models. Several teams have explored global approaches to modulate the amount of DNA methylation in animals (Ryley Parrish et al., 2013; Williams-Karnesky et al., 2013). Results have been conflicting to date, however, with both increased and decreased excitability being reported. What may be needed is a context-specific or targeted approach to increase or decrease DNA methylation. Elegant experimental studies in cells and animal models using the genome editing tool, CRISPR (clustered regularly interspaced short palindromic repeats), recently demonstrated this was possible. Researchers tagged an enzyme that either increased or decreased DNA methylation onto a CRISPR variant and directed it to specific sites in the genome whereupon the epigenetic mark was modified (Liu et al., 2016). If critical genes are epigenetically silenced in epilepsy, this type of approach could be considered. For example, the gene encoding Reelin is hypermethylated in epilepsy patients with granule cell dispersion (Kobow et al., 2009). Re-activating the gene could be an approach to restore secretion of this protein and perhaps arrest granule cell layer dispersion. Genome-wide methylation mapping of the hippocampus in patients with temporal lobe epilepsy identified multiple hyper-methylated genes (Miller-Delaney et al., 2015). Although few had biologically plausible links to epilepsy, they (or others) might be targets for this type of selective approach in the future. This is also seen in brain tissue from various animal models of acquired epilepsy (Kobow et al., 2013; Debski et al., 2016).
Targeting of specific non-coding RNAs is another therapeutic approach that might be tried. Non-coding RNAs can be manipulated using antisense oligonucleotides (OGNs). These can be designed to be extremely potent and selective. CUR-1916 is the first OGN targeting a non-coding RNA to reach clinical trials for epilepsy. The OGN targets the natural antisense transcript that is produced by SCN1A which encodes the sodium channel that is most often mutated in patients with Dravet syndrome (Hsiao et al., 2016). By blocking the SCN1A-NAT, CUR-1916 is expected to upregulate the levels of the SCN1A transcript. Given that the majority of mutations cause loss-of-function in epilepsy (and other neurological diseases), this therapeutic approach has particular promise. The main problem with all OGNs, however, is delivery. They cannot pass from the circulation into the brain as they are too large to cross the [intact] blood-brain barrier. They must therefore be injected intrathecally. The results of trials of this and other OGNs are eagerly anticipated.
There are a number of important challenges with manipulating epigenetic processes that may limit clinical translation. As mentioned in the preceding text, the machinery for epigenetic change is present in all cell types, therefore cell-specific manipulations may need to be developed so that epigenetic marks are changed in the right cell. Expression of epigenetic components is much higher in dividing cells, therefore cells within the neurogenic niche in the brain (e.g. the subgranular zone of the dentate gyrus) may be affected most. Then there are timing aspects. Aging is associated with a trend toward a closed transcriptional state (Berson et al., 2018). Conversely, the developing brain may be highly susceptible to epigenetic manipulations, which creates opportunities but also risks. Finally, epigenetic systems are excellent at storing memories and brain tissue may retain a form of memory for seizure activity. We may need to “wipe clean” epigenetic marks in order to get better outcomes. For example, to prevent previously epileptogenic tissue from re-starting hyper-excitable behaviour following resective surgery for drug-resistant epilepsy.
Summary, prospects, challenges and next steps
Epigenetic processes shape the gene expression landscape by controlling access to the genetic code. It is increasingly important that we think about epigenetics when considering mechanisms controlling gene expression. There is growing evidence that this process is disturbed following epileptogenic injuries, and some of the recently discovered epilepsy genes encode proteins that perform epigenetic functions. Epigenetic marks persist in the chronic epileptic state, perhaps stabilizing aberrant gene expression. Epigenetic marks may serve as diagnostic biomarkers, and epigenetic processes lend themselves to therapeutic targeting. However, the bulk of data in our field has come from animal models which may not extrapolate to human epilepsies.
How do we see the field progressing and where are the opportunities? (Box 3: Outstanding Research Questions). Technology will continue to drive discoveries in this area. Elucidating the epigenome of epilepsy is a key challenge. We need more solid data on each of the major epigenetic processes in experimental and human epilepsy. Combining deep sequencing with proteomics and other emerging technologies, such as single-cell sequencing, could provide us with the epigenome at individual cell resolution. Beyond being a scientific achievement, this will help us to see where therapies could be deployed, for example to “re-activate” a silenced gene or silence pathogenic genes. It may indicate a need to direct therapies toward specific cells types. Alongside precision epigenetic therapies, there may still be a place for broad-acting epigenetic drugs which have yet to be tested in clinical trials. It is likely that many more non-coding RNAs exist that regulate transcription and chromatin structure, and these may harbour new targets in the future. Achieving these aims will require multi-disciplinary approaches. Last, what is the role of epigenetic processes in presumed genetic epilepsies and could correction of these be a novel approach to therapy? The results should provide a better understanding of how gene expression is altered in epilepsy and may be corrected, with the development of innovative molecular diagnostics and therapeutics.
Author contributions
DCH wrote the manuscript with KK, CAR and EAvV. AJB, AMG, GLC, SH, IL-C, HMK, AP and MRJ contributed to concepts or literature analyses, and read and approved the final manuscript.
Acknowledgements and disclosures
This report was written by experts selected by the International League Against Epilepsy (ILAE) and the American Epilepsy Society (AES) and was approved for publication by the ILAE and the AES. Opinions expressed by the authors, however, do not necessarily represent the policy or position of the ILAE or the AES. This report is a product of the ILAE/AES Genetics/Epigenetics Task Force.
DCH reports research support from the European Union's “Seventh Framework” Programme (FP7) under Grant Agreement no. 602130 (EpimiRNA), Science Foundation Ireland (SFI) under grants SFI/13/IA/1891, SFI/14/ADV/RC2721, and 16/RC/3948, and co-funding under the European Regional Development Fund and by FutureNeuro industry partners. KK reports research support from the European Union's ‘Seventh Framework’ Programme (FP7) under Grant Agreement no. 602531 (DESIRE). CAR reports support from the National Health and Medical Research Council (NHMRC) Program Grant (10915693). EAvV has received funding from the European Union's “Seventh Framework” Programme (FP7) under Grant Agreement no. 602102 (EPITARGET) and is supported by the PPP Allowance made available by Top Sector Life Sciences & Health to the Dutch Epilepsy Foundation to stimulate public-private partnerships.