Hématologie
MENUL’hématopoïèse cyclique, un phénotype, deux gènes différents. Les modalités d’une interaction Volume 10, numéro 3, Mai-Juin

- Page(s) : 245-7
- Année de parution : 2004
Auteur(s) : Dominique Labie
Institut Cochin, GDPM, 24, rue du Faubourg Saint-Jacques, 75014 Paris, France
L’hématopoïèse cyclique humaine est une maladie autosomique dominante, caractérisée par une oscillation périodique d’environ 21 jours de production des cellules sanguines. Les neutrophiles, en particulier, oscillent entre zéro et des valeurs presque normales, permettant entre temps le développement d’infections opportunistes. Le criblage du génome, et un clonage positionnel, ont permis en 1999 à une équipe de l’Université de Washington, Seattle, États-Unis, de localiser le gène responsable en 19p13.3 et de mettre en évidence chez des malades sept substitutions du gène ELA2, codant pour une sérine protéase de 240 aa. normalement présente dans les granules des neutrophiles et des monocytes [1]. Cette observation a été corroborée dans une autre étude, évoquant le rôle de la même élastase dans les neutropénies congénitales sévères (SCN), les mutations semblant cependant différentes dans les deux syndromes [2]. Il s’agît, dans la majorité des cas, de mutations de novo, l’enzyme mutée interférant avec le trafic normal [3].Un phénotype comparable, autosomique récessif, d’hématopoïèse cyclique a été observé chez le chien, mais aucune mutation du gène homologue ELA2 n’a été retrouvée [4]. Chez le chien la maladie s’accompagne de plaques d’hypopigmentation qui évoquent le syndrome d’Hermansky-Pudlak chez l’homme. Les études moléculaires, dans ces derniers cas, avaient mis en évidence des mutations de la sous-unité β3a du complexe adaptateur AP-3 [5]. Ce complexe est un hétérotétramère (deux dimères α3/δ et µ3/β3) associé au trans-Golgi, et qui serait impliqué dans le transport des protéines cargo vers les lysosomes [6, 7]. Des mutations de la sous-unité Ap3b1 ont aussi été mises en évidence dans le modèle murin de la maladie d’Hermansky-Pudlak, entraînant une dégradation globale de AP-3 [8]. Une autre étude, comparant chez la souris les mutants naturels et ceux obtenus par invalidation génique, a démontré que le gène Ap3b1 est bien à l’origine du phénotype, par suite d’un défaut de transport des protéines membranaires [9].
L’ensemble de ces données a amené les auteurs du dernier article [4] à explorer le gène AP3B1 au cours de l’hématopoïèse cyclique du chien. Ils ont ainsi mis en évidence l’insertion d’une adénine dans une séquence 9A de l’exon 20, traduite par un décalage de phase de lecture et une terminaison prématurée de la traduction. Au niveau protéique, on ne retrouvait ni la sous-unité β3a mutée, ni la sous-unité µ3a dégradée.
Des mutations dans des gènes différents entraînant un phénotype similaire évoquent évidemment que les produits de ces gènes interagissent fonctionnellement, et c’est ce qui a été recherché. L’hypothèse était que AP-3 reconnaisse l’élastase neutrophile comme une protéine cargo et assure son transport vers les granules. Le mode d’interaction a été exploré en technique de double hybride : avec d’une part les sous-unités β3a ou µ3a de AP-3, d’autre part la moitié distale de l’élastase neutrophile, ou un variant. L’élastase neutrophile comporte, en effet, une extension C-terminale de 20 résidus, clivable par protéolyse, de fonction jusqu’alors inconnue. L’expérience a été fait avec ou sans cette extension. On a constaté que la protéine clivée interagît avec µ3a, mais pas avec β3a, la protéine non clivée n’interagissant avec aucune des deux sous-unités. L’interaction se présentait comme inhibée par la présence de l’extension C-terminale. L’examen de la séquence protéique, par ailleurs, ne montre qu’une seule tyrosine en position 199 (séquence LYPDA). Cette tyrosine devait être le signal de sortie et d’interaction avec µ3a. L’exploration par algorithmes de la séquence de l’élastase a montré l’existence d’un domaine transmembranaire juste avant ce signal Y. Les mutations les plus fréquentes s’alignent dans ce domaine (figure 1). En délétant ce signal Y, sont-elles susceptibles de modifier le trafic subcellulaire ?
Plusieurs abords ont été utilisés pour vérifier cette hypothèse. Elles ont utilisé, par comparaison avec l’élastase neutrophile normale, deux constructions de mutants : La mutation 205ΔC correspond à une délétion de l’extension C-terminale et retient le signal de sortie, la mutation Y199X abolit ce signal. Les différentes protéines ont été localisées par immunofluorescence dans des cellules basophiles de rat dénuées d’élastase endogène. La forme sauvage se localise majoritairement dans les granules, un peu dans la zone périnucléaire, la localisation granulaire s’accentue chez le mutant 205ΔC, alors que l’élastase n’est retrouvée qu’au niveau de la membrane plasmique et de l’enveloppe nucléaire quand il y a suppression du signal chez Y199X. Un autre contrôle a utilisé les mêmes constructions explorées par Western blot avec des résultats comparables. La question se posait de savoir comment peut s’opérer cette interaction. L’élastase est une protéine soluble qu’on trouve dans les granules, alors que AP-3 revêt la surface cytoplasmique des vésicules. Les deux protéines sont situées sur les surfaces opposées d’une membrane. L’interaction requiert donc que l’élastase traverse la membrane. Les expériences ont comparé l’élastase sauvage normale à deux mutants naturels, dont l’un était une délétion partielle du domaine transmembranaire (ΔV161-F170), l’autre un variant R191Q qui augmentait l’hydrophobicité de la protéine. Elles ont comporté d’une part des ultracentrifugation de lysats cellulaires séparant la fraction soluble et la fraction membranaire, d’autre part le traitement des neutrophiles par un certain nombre d’agressions, perméabilisation par congélation/décongélation, protéolyse par la protéinase K, traitement par le triton, éventuellement suivie de la localisation des différentes protéines par des anticorps spécifiques.
Les auteurs proposent une explication de l’ensemble des résultats obtenus. L’élastase neutrophile existe normalement sous deux isoformes de 34 et 31 kDa, avec ou sans l’extension C-terminale. L’ultracentrifugation sépare la fraction de 31 kDa soluble granulaire de la fraction de 34 kDa insoluble et membranaire. La maturation normale implique que la forme qui a retenu le C-terminal traverse les membranes du trans-Golgi vers le cytosol. Après clivage du C-terminal, la protéine, associée à la membrane, fixe AP-3. La protéine clivée est alors libérée dans le lumen et transportée vers les granules, la perte du signal ou l’absence de clivage empêchant de fait l’interaction. La localisation vers les membranes est due à un routage erroné. Dans l’hématopoïèse cyclique du chien, il y a déficit en AP-3, alors que dans la plupart des cas de neutropénie congénitale sévère il y a délétion du signal, mais l’erreur de routage est la même. Dans l’hématopoïèse cyclique humaine, en revanche, les mutations entraînent le plus souvent une interruption du domaine transmembranaire et la protéine soluble se localise dans les granules.
Références
1. Horwitz M, Benson KF, Person RE, Aprikyan AG, Dale DC. Mutations in ELA2, encoding neutrophil elastase, define a 21-day biological clock in cyclic haematopoiesis. Nature Genet 1999 ; 23 : 433-6.
2. Dale DC, Person RE, Bolyard AA, Aprikyan AG, Bos C, Bonilla MA, Boxer LA, Kannourakis G, Zeidler C, Welte K, Benson KF, Horwitz M. Mutations in the gene encoding neutrophil elastase in congenital and cyclic neutropenia. Blood 2000 ; 96 : 2317-22.
3. Li FQ, Horwitz M. Characterization of mutant neutrophil elastase in severe congenital neutropenia. J Biol Chem 2001 ; 276 : 14230-41.
4. Benson KF, Li FQ, Person RE, Albani D, Duan Z, Wechsler J, Meade-White K, Williams K, Acland GM, Niemeyer G, Lothrop CD, Horwitz M. Mutations associated with neutropenia in dogs and humans disrupt intracellular transport of neutrophil elastase. Nature Genet 2003 ; 35 : 90-6.
5. Dell’Angelica EC, Shotelersuk V, Aguilar RC, Gahl WA,Bonifacino JS. Alterd trfficking of lysosomal proteins in Hermansky-Pudlak syndrome due to mutations in the β3a subunit of the AP-3 adaptor. Molec Cell 1999 ; 3 : 11-21.
6. Simpson F, Peden AA, Christopoulou L, Robinson MS. Characterization of the adaptor-related protein complex, AP-3. J Cell Biol 1997 ; 137 : 835-45.
7. Robinson MS, Bonifacino JS. Adaptor-related proteins. Curr Op Cell Biol 2001 ; 13 : 444-53.
8. Feng L, Seymour AB, Jiang S, To A, Peden AA, Novak EK, Zhen L, Rusiniak ME, Eicher EM, Robinson MS, Gorin MB, Swank RT. The β3A subunit gene (Ap3b1) of the AP-3 adaptor complex is altered in the mouse hypopigmentation mutant pearl, a model for Hermansky-Pudlak syndrome and night blindness. Hum Mol Genet 1999 ; 8 : 323-30.
9. Yang W, Li C, Ward DM, Kaplan J, Mansour SL ; Defective organellar membrane protein trafficking in Ap3b1- deficient cells. J Cell Sci 2000 ; 113 : 4077-86.
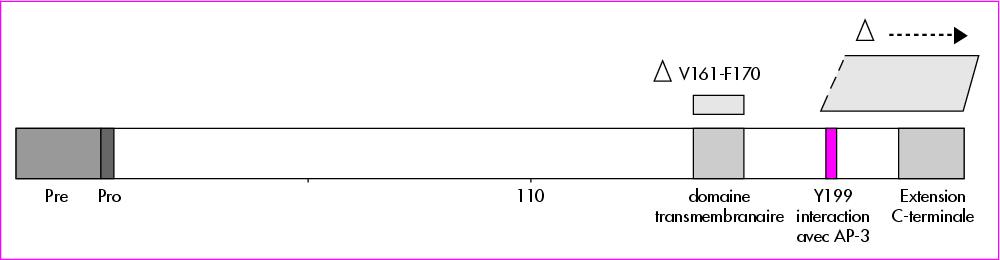
Figure 1. Représentation schématique de l’élastase neutrophile. En N-terminal les séquences pré- et proélastse. En C-terminal l’extension de 20 aa qui sera clivée par protéolyse. Les mutations ponctuelles sont réparties sur l’ensemble de la séquence (cf. réf. 4), elles ne sont pas figurées ici. Deux catégories de mutations sont représentées spécifiquement. 1) La délétion ΔV161-F170 est retrouvée de façon itérative, entraînant presque toujours une neutropénie cyclique. Elle représente une interruption partielle du domaine transmembranaire, le signal Y199 est respecté. Le routage de l’élastase, soluble, se fait vers les granules. 2) Une série de mutations, à partir du résidu 192 et au-delà, entraîne, par décalage de phase de lecture, une terminaison prématurée de la traduction avec suppression du signal et délétion de toute l’extrémité C-terminale. Ces mutations sont à l’origine d’une neutropénie congénitale sévère et d’un routage erroné de l’élastase vers les membranes.bn