Epileptic Disorders
MENUManaging Lafora body disease with vagal nerve stimulation Volume 19, numéro 1, March 2017

- Mots-clés : vagal nerve stimulation, Lafora body disease, progressive myoclonus epilepsy
- DOI : 10.1684/epd.2017.0892
- Page(s) : 82-6
- Année de parution : 2017
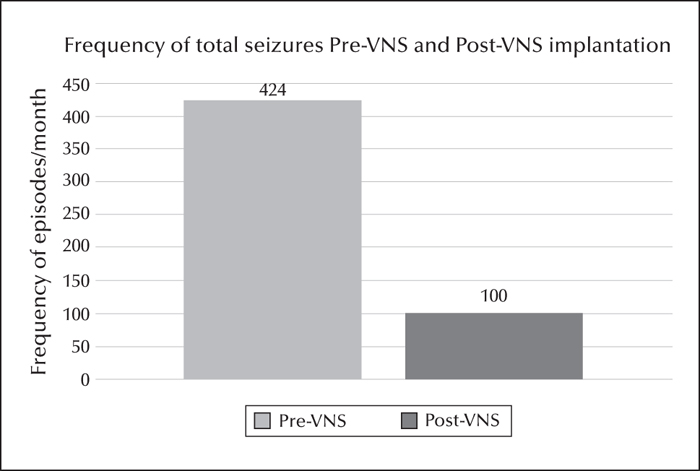
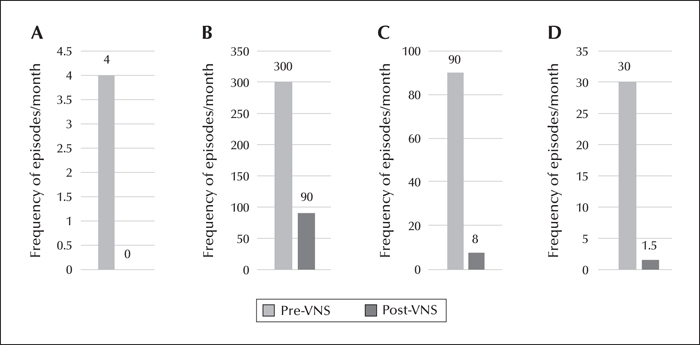
A 17-year-old female, of consanguineous parents, presented with a history of seizures and cognitive decline since the age of 12 years. She had absence, focal dyscognitive, generalized myoclonic, and generalized tonic-clonic seizures, all of which were drug resistant. The diagnosis of Lafora body disease was made based on a compatible clinical, EEG, seizure semiology picture and a disease-causing homozygous mutation in the EPM2A gene. A vagus nerve stimulator (VNS) was inserted and well tolerated with a steady decrease and then stabilization in seizure frequency during the six months following insertion (months 1-6). At follow-up, at 12 months after VNS insertion, there was a persistent improvement. Seizure frequency during months 7-12, compared to pre-VNS, was documented as follows: the absence seizures observed by the family had decreased from four episodes per month to 0 per month, the focal dyscognitive seizures from 300 episodes per month to 90 per month, the generalized myoclonic seizures from 90 clusters per month to eight per month, and the generalized tonic-clonic seizures from 30 episodes per month to 1.5 per month on average. To our knowledge, this is the second case reported in the literature showing efficacy of VNS in the management of seizures in Lafora body disease.