Epileptic Disorders
MENUSensory stimulus-sensitive drop attacks and basal ganglia calcification: new findings in a patient with FOLR1 deficiency Volume 16, issue 1, March 2014





5-Methyltetrahydrofolate (5-MTHF) is the biologically active form of folate and is essential for DNA repair and synthesis, as well as homocysteine degradation. 5-MTHF is transported into the CSF, particularly at the choroid plexus, where the folate receptor alpha (FRα) is anchored (Hyland et al., 2010; Mangold et al., 2011). Mutations in the folate receptor 1 gene (FOLR1) are associated with normal plasma folate but severely reduced concentrations of 5-MTHF in the CSF. This indicates that the gene product, folate receptor alpha (FRα), plays a crucial role in the transport process across the blood brain barrier (Steinfeld et al., 2009; Grapp et al., 2012). Affected patients share an extremely low 5-MTHF concentration in the CSF, delayed myelination or hypomyelination on MRI, and suffer from developmental regression, ataxia, and myoclonic seizures or drop attacks (Pérez-Dueñas et al., 2010; Grapp et al., 2012). Here, we report on a further patient with FOLR1 deficiency and describe the unusual clinical finding of triggered drop attacks and bilateral spot calcification within the basal ganglia.
Case report
This 5-year-old boy has a healthy older half-brother and half-sister. The family originated from Ghana. Consanguinity was denied. The boy was born at term. His birth weight was 3,210 g (P10-25), length 51 cm (P25-50), and head circumference 33 cm (P3). Until the age of about 2 years, his psychomotor development was unremarkable. Independent walking was achieved at the age of 10 months and first words at 12 months. Since the age of 2 years and 6 months, hyperactive behaviour was noticed. At 3 years of age, he was admitted to the hospital for diagnostic work-up, for marked generalised ataxia and intellectual disability (developmental quotient: 0.5), that did not reveal a specific diagnosis. At the age of 4 years and 3 months, he was readmitted due to a single generalised epileptic seizure. Thereafter, he suffered from daily sudden drop attacks, which were to some extent triggered by actions associated with body care, blowing his nose or washing his face or hands, and upon passive eye closure during EEG. Attacks were accompanied by a rapid elevation and extension of his arms and a flexion of neck and trunk. These myoclonic-tonic events lead to several facial injuries despite preserved consciousness (video sequence 1 and 2). Occasionally, the attacks occurred in short series of two or three. In addition, he had frequent episodes with head stereotypies associated with abnormal eye deviation to the right side (video sequence 3), multifocal myoclonia, and atonic head drops. These events were classified as non-epileptic, as EEG was normal during these episodes. Antiepileptic treatment with valproic acid, ethosuximide, vigabatrin, levetiracetam, and clonazepam were of no clear benefit. Myoclonia and gait ataxia worsened while on vigabatrin. His cognitive and physical skills deteriorated with progressive gait ataxia, generalised choreic movements, myoclonia, irritability, limited use of language, and progressive microcephaly (1.3 cm <P3).
EEG
At the beginning of the myoclonic-tonic drop attacks, interictal EEG was normal. Spontaneous and provoked drop attacks (i.e. by eye closure; video sequence 4) were not associated with ictal EEG features, however, interpretation of the EEG was limited by distinct movement artefacts during the myoclonic-tonic event. During follow-up (six months), interictal EEG showed an impressive deterioration with high-voltage spike and sharp wave activity and a slow high-amplitude background activity (figure 1).
Laboratory work-up
Initial metabolic work-up was normal apart from persistent hypochromic anaemia due to alpha thalassaemia minor. 5-MTHF in the CSF was below the detection limit (<0.0 nmol/L; normal range: 41-117 nmol/L) (Blau and Thöny, 2008). Sequencing of all coding exons and exon-intron boundaries of the FOLR1 gene (gene ID ENSG00000110195, transcript/cDNA ID ENST00000312293.3; NM_016725.1) revealed homozygosity for the known nonsense mutation c.610C>T, p.Arg204* in exon 5 (Dill et al., 2011).
Imaging
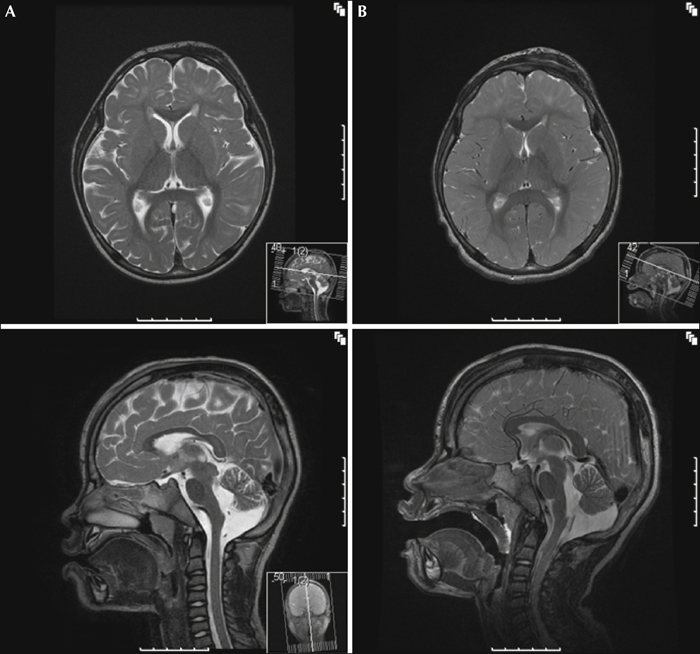
Cranial MRI obtained at the age of 3 years and 2 months revealed generalised hypomyelination, with lack of progress at repeat MRI at the age of 4 years and 10 months (figure 2A). In addition, cerebellar atrophy became more apparent. MR spectroscopy revealed reduced peaks of myoinositol and choline within the parietal white matter and the basal ganglia. After five months of treatment with folinic acid, MRI showed impressive gain of brain volume in both hemispheres and the cerebellum (figure 2B). Computed tomography (CT), performed due to head trauma at the age of 5 years and 2 months, revealed bilateral calcifications within the basal ganglia (figure 3), not visible on both MRI series.
Course of treatment
At the age of 5 years, treatment with folinic acid (leucovorin) was started at a dose of 4.5 mg/kg body weight/day orally and administration of 90 mg folinic acid intravenously (iv) every 1-2 weeks. After one month, the boy was less irritable and gait ataxia improved. His behaviour and attention improved, head stereotypes disappeared, but myoclonic-tonic drop attacks persisted. Due to the continued low level of 5-MTHF in CSF (20.1 nmol/L; normal range: 41-117 nmol/L), folinic acid substitution was increased to 5.6 mg/kg/body weight/day orally with weekly administration of 120 mg of folinic acid iv over 1-2 hours for three months (5-MTHF in CSF: 33 nmol/L). Over a period of four months of therapy, this led to a significant 50-75% reduction of his drop attacks. Ataxia of the extremities and myoclonic jerks persisted. The EEG remained abnormal with high-voltage spike and sharp wave activity. Based on a case report of a patient carrying the same mutation (Dill et al., 2011), a five-week trial with pyridoxine at 200 mg/day (10 mg/kg), followed by a three-week trial with pyridoxal 5’-phosphate at 300 mg/day (15 mg/kg), was performed without clinical benefit.
Discussion
Diagnostic work-up of epileptic encephalopathies is based on careful delineation of seizure semiology and accompanying findings. Myoclonic attacks, falls, and spasms appear to be typical of patients with FOLR1 mutation. The most impressive clinical symptoms in our patient were the distinct myoclonic-tonic drop attacks, partly provoked by touching the face, hand washing, or emotional stress. This led to the initial assumption of stimulus-induced drop attacks. In addition, the patient exhibited a variety of symptoms consisting of myoclonic jerks, atonic head drops, abnormal eye movements, and grimacing, all of which were not associated with any ictal EEG activity. A similar phenotype and seizures reminiscent of infantile spasms have also been described in a larger case series of 11 patients with FOLR1 mutations (Grapp et al., 2012). These patients had a slow background rhythm with multifocal epileptiform activity on EEG, while ictal EEG changes were not described in this cohort. The patient published by Pérez-Dueñas et al. (2010) suffered from daily tonic and myoclonic attacks. The EEG showed slow high-amplitude background activity, frequent myoclonic jerks, and tonic seizures. The multifocal myoclonic jerks on electromyographic recording (deltoids) were not associated with EEG activity.
Frequent multifocal and generalised myoclonic jerks, intermixed with tonic seizures, spasms, abnormal eye movement, grimacing, or irritability, are prominent symptoms in neonates with pyridoxine-dependent epilepsy (PDE) and pyridoxine phosphate oxidase deficiency (PNPO) (Schmitt et al., 2010). Also, in these two metabolic epileptic encephalopathies, paroxysmal events are not necessarily of epileptic origin (Schmitt et al., 2010). The lack of conclusive ictal EEG pattern and the reproducible trigger mechanisms in our patient led us to classify these events as seizures with caution. On the other hand, we observed highly abnormal interictal EEG with spike and wave activity, compatible with an epileptic encephalopathy and seizures.
In contrast to the patient with FOLR1 mutation reported by Dill et al., our patient did not benefit from additional pyridoxine and pyridoxal 5’-phosphate treatment.
This is the first report of the presence of basal ganglia calcifications in a patient with FOLR1 deficiency. Though basal ganglia calcifications lack phenotypic specificity (Livingston et al., 2013), the fact that they are also present in autosomal recessive hereditary folate malabsorption (MIM # 229050) points towards a causal relationship with cerebral 5-MTHF deficiency.
Conclusion
For young children with ataxia and seizures, that may resemble spasms or myoclonic astatic attacks, in combination with delayed brain myelination, the CSF should be tested to determine 5-MTHF deficiency. This case expands the clinical spectrum of patients with FOLR1 mutation to include provoked myoclonic-tonic attacks that are not associated with ictal EEG patterns, which may precede the characteristic interictal EEG pattern of slow high-amplitude background rhythm with multifocal epileptiform activity.
Response to treatment with folinic acid depends on the age of the patient and duration of symptoms, thus, early diagnosis appears to be crucial in order to prevent irreversible cognitive deficits. Calcifications within the basal ganglia might be a further diagnostic clue, which has not been reported so far, probably due to the current marked preference of MRI over CT.
Acknowledgements and disclosures
We thank the mother of the patient who allowed us to publish the videos of her son, the nursery teachers of Imago preschool for providing us with the video material, Prof R Steinfeld for handing out advice concerning treatment, and Corinne Britschgi and Anahita Rassi for technical assistance at the Division of Clinical Chemistry and Biochemistry.
None of the authors has any conflict of interests to disclose.